Topics
2025.03.31 circular No.24
金属電池とイオン液体京都大学大学院エネルギー科学研究科
松本 一彦
二次電池のようなエネルギー貯蔵デバイスの電解液にイオン液体(IL)を用いるというのは、私が学生になる前から研究され続けてきた分野で1,2、イオン液体の応用としては最も古いものの一つといえる(図1)3。歴史を紐解くと、1990年代にフッ素が入った安定性の高いアニオンが使われ始めて、イオン液体は広く用いられるようになったとされているが2,4,5、非水系の電池はそもそも空気中に出さないので、使い道がはっきりしていれば、多少空気中で不安定であっても利用できる可能性はある。二次電池への応用といえば、リチウムイオン電池がまず思い浮かぶわけだが、下に述べる通り、イオンの挿入脱離を利用するイオン電池より、単純な金属の析出溶解を利用する金属電池の方が貯蔵できるエネルギーは大きい。本稿ではイオン輸送などバルク特性ではなく、界面特性に注目して、金属電池にイオン液体電解質を用いる優位性と関連研究について、筆者らの取り組みも含めて紹介する。
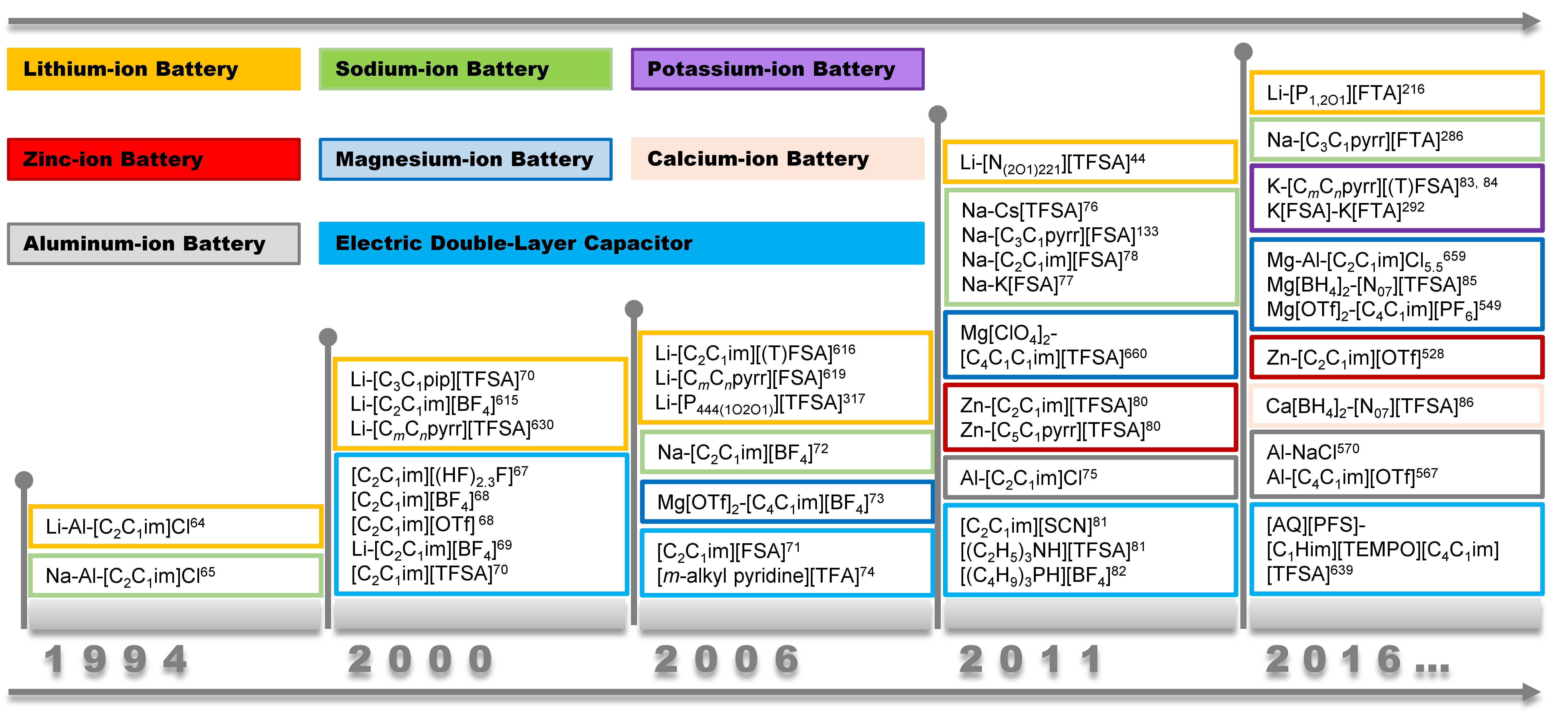
図1 エネルギー貯蔵デバイス用イオン液体電解質の開発年表.エルセビア社より許可を得て転載.3
2.金属電池とは
現在エネルギー貯蔵デバイスの分野で単に金属電池というと、充放電ができる金属二次電池を指すことが多く、放電しかできない金属一次電池とは区別して考えることが多い。実は我々の身の回りにある一次電池(使い切りの電池)は一般的に負極反応が金属溶解であるため、金属一次電池といえるが、わざわざこのように呼ぶことはあまりない。例えば水溶液系のマンガン電池やアルカリ電池は亜鉛の酸化的溶解が負極反応であり、非水溶液系のリチウム電池はリチウムの酸化的溶解が負極反応である(リチウム金属は水と反応するため、有機溶媒を用いて商用化されている)。いずれも二次電池化が期待されているが、それが容易でないことは広く認識されている。
なぜ金属電池(以下金属二次電池の意味で用いる)が注目されているかというと、金属負極の単位重量あるいは単位体積当たりに貯蔵できる電気量(一般的に容量と呼ばれ、単位はmAh g−1またはmAh cm−3)が高いからである。例えば図2でリチウム金属電池とリチウムイオン電池を比較する。現行のリチウムイオン電池は負極にグラファイトが用いられており、その層状構造の隙間にリチウムイオンが挿入・脱離することで充放電が進行する(グラファイトはホスト材料と呼ばれる)。この時、どんなに詰め込んでも炭素六個当たりリチウム一個しか吸蔵できないので、ホスト材料のグラファイトの重量による制約からは逃れられず、理論容量は372 mAh g−1となる(式(1))。
Li+ + e− + 6C = LiC6 |
(1) |
負極がリチウム金属の場合、充放電反応は単純な金属リチウムの析出溶解である(式(2))。
Li+ + e− = Li |
(2) |
この場合ホスト材料が無いので、グラファイトのようにホスト材料あたりの容量(放電状態の容量)を計算することはできず、リチウム金属あたりの容量(充電状態の容量)で議論される。リチウムは原子量が小さいため、単位重量当たりの理論容量は3860 mAh g−1と非常に高く、これがリチウム金属電池を魅力的にする要因である。なお、この理論容量を上記のグラファイトの理論容量と比較するのは不適切で、同じく充電状態にあるLiC6を基準とした339 mAh g−1と比較するべきである。
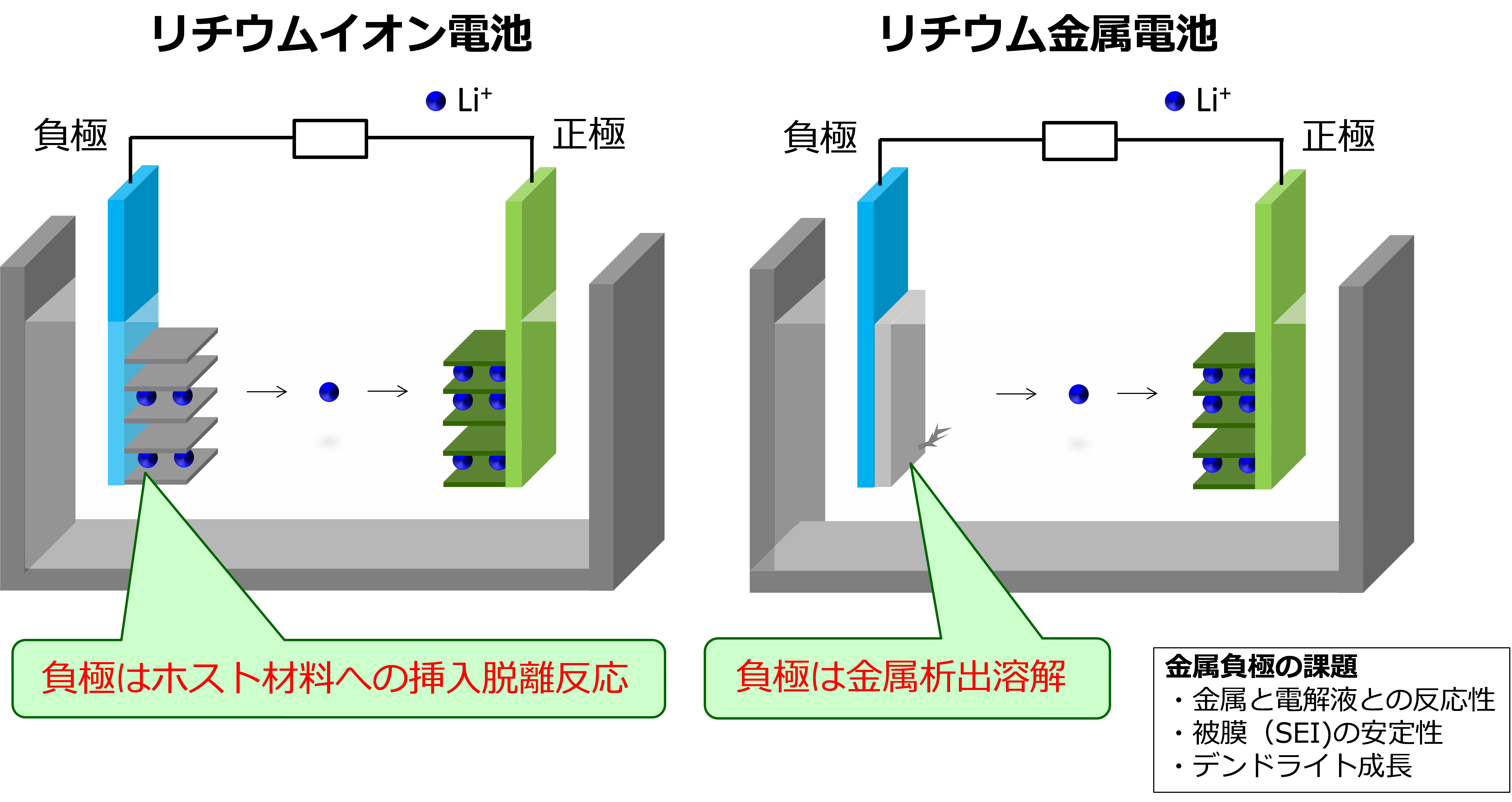
図2 リチウムイオン電池とリチウム金属電池の違い.金属負極の課題も合わせて示す.
金属電池を考える上で、セル構成の理解が重要であるため、初期状態におけるリチウム金属の配置から三つの代表的な構成に分けて、図3にまとめた。(1)はハーフセルと呼ばれる構成で正極(あるいは正極と見立てた負極)の性能評価に利用される。リチウム金属は過剰に負極に載せられており、これは金属電池とは呼ばない(あくまで電極材料評価の対極としてリチウム金属が利用されている)。(2)はアノードフリーと呼ばれる構成で、最も高い容量を与える配置であるが(理論容量も通常この構成で計算されている)、過剰なリチウム金属が負極に存在しないため、サイクル特性が悪く、実現が難しい。(3)は初期状態で所定量のリチウム金属を負極に載せてある構成であり、アノードフリーの場合より余分にリチウム金属がある分、サイクル特性に優れる。(1)と異なり(3)では負極に載せるリチウム金属の量を制御することが重要なポイントとなる(この量で電池のエネルギー密度が決まる)。容量劣化がなければ(2)が理想的だが、現実的には(3)も含めて電池構成が考えられているのが実情である。6

図3 初期状態における金属電池の構成に関する模式図.(1)ハーフセル、(2)金属電池(I)、(3)金属電池(II).例としてリチウム金属電池の場合を示している.セパレータは省略してある.
リチウムイオン電池と比較して、リチウム金属電池が広く実用化していない理由は、式(2)で示されるリチウム金属の析出溶解が難しいからである7−9。Li+/Liの標準酸化還元電位は標準水素電極基準で−3.0 Vと低く、そのおかげで高エネルギー密度の電池を作ることができるのだが、このような還元雰囲気で安定な電解液はなかなか存在しない。グラファイト負極の場合SEIと呼ばれる表面被膜が電極表面に形成することで電解液の連続的な還元分解を防いでいるが、リチウム金属負極の場合、充放電するたびに金属が析出溶解して新しい表面が生成するため、電解液の還元分解を防ぐことが難しい。電解液の還元分解は通常初期サイクルで最も大きいが、仮に99 %の平均析出溶解効率で充放電できても、電池の容量劣化は激しい。一日一回完全に充電して、完全に放電とすると、一年で0.99365 < 0.026となり初期容量の3%も残っていない。もう一つの問題はリチウム金属が析出する際に平滑に(基板に密着して平らに)析出せず、樹枝状に(とげとげに)析出することである(デンドライト形成という)。樹枝状に金属が析出すると、どこかでポロっと落ちてしまい、導通がなくなるため、溶解できず容量が落ちてしまう(デッドリチウムと呼ばれる)。また、樹枝状のリチウム金属は負極からどんどん成長して、正極と負極を電気的に絶縁しているセパレータも突き抜け、最後には正極に到達してショートを引き起こす。これは電池の発火につながる危険な現象で、安全上大きな課題である。
このように問題山積の金属電池ではあるが、近年技術的な進歩が著しく、実用化の可能性が高まっていると感じている。これは研究者らの様々な努力の積み重ねによることはいうまでもないが、電解液の進歩によるところが大きい。次節以降では金属電池の開発においてイオン液体が果たす役割を述べる。
イオン液体は還元耐性があまり高くないため、リチウムイオン電池の電解液として使用するとリチウム金属に対する安定性が低く、またグラファイト負極ではグラファイトの剥離や低電位での分解が起こるという課題があった。この課題を解決したのは、ビス(フルオロスルフォニル)アミド(N(SO2F)2−: FSA−)塩であり(少し古いが、フルオロスルフォニルアミド塩の総説があるのでご興味があればご参照いただきたい10)、2006年に産総研と関西大学の両グループからFSAイオン液体のリチウムイオン電池への応用が報告された(学術的には日本発!)11,12。これらの報告ではハーフセル試験におけるLiCoO2正極とグラファイト負極の安定な作動について述べられているが、いずれも他のイオン液体の場合と比較して、安定なリチウム金属対極の作動が実は重要なポイントである(金属リチウム対極が正常に作動しないとコインセル試験が続かない)。なお、当初はLi[FSA]の入手が難しく、リチウム塩はLi[TFSA]など他のアニオンの形で導入されていたケースが多い。
イオン液体を用いたリチウム金属電池の文献は多くあるので、ここではいくつかキーとなるものを紹介する。FSAイオン液体も金属リチウムと反応するが、安定なSEI(solid-electrolyte interphase)が形成されるので、長期的な電気化学特性に優れるとされる。ここで、FSA−の還元分解機構について触れておきたいと思う。2014年の報告によると、FSA−の放射線分解による生成物を用いた電子スピン共鳴分光で、下記の反応式に基いた安定な∙SO2NSO2F−ラジカルイオンの生成が指摘されている13。
Li[N(SO2F2)2]+e−→LiF+ ∙SO2NSO2F− |
(3) |
このような安定なラジカルイオンは、ビス(トリフルオロメチルスルフォニル)アミド(N(SO2CF3)2−: TFSA−)の分解では見られない。また、∙SO2NSO2F−はSO2ガスの放出や有機カチオンと反応が起こりづらいため、これが化学的に分解されることなく、さらに還元されてSO2F−とNSO2−が生成するとされ(Li[SO2F]の安定性には個人的に疑問がある)、結果的に形成される無機物を中心としたSEIは安定であるというのがここで述べられている機構である(図4)。2018年の論文では、核磁気共鳴分光、X線光電子分光、赤外分光、電気化学インピーダンス分光を組み合わせて、さらに詳細な議論がなされている14。この報告では、Li[FSA]-[P111i4][FSA](P111i4+ = trimethyl(isobutyl)phosphonium)イオン液体中で形成されたSEIを内部層と外部層に分けて考えており、内部層はFSA−由来の無機的な成分(LiF、Li2O、Li[SO2]、Li[NSO2F](Li2CO3については不確定))で、外部層は有機カチオン由来の有機成分(還元されていない可能性もある)からなると述べている。また、上述の2014年の文献とは異なり、∙SO2NSO2F−を経由せず、FSA−の還元生成物であるFSO2NSO2F2−から直接N−S結合が解離する機構もこの文献では提案されている。
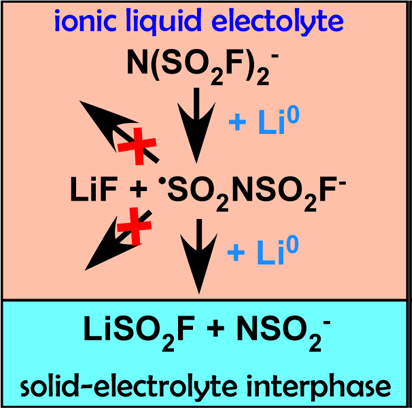
図4 FSA−アニオンの還元分解機構の概略図.アメリカ化学会より許可を得て転載.13
次にイオン液体を用いたリチウム金属電池の電気化学特性についていくつか紹介する。2016年の文献では、リチウム金属をLi[FSA]-[C3C1pyrr][FSA](C3C1pyrr: N-methyl-N-propylpyrrolidinium)イオン液体に12日以上浸漬しておくと安定なSEIが形成し、デンドライト成長が抑制され、長期間安定して作動するリチウム金属電池を実現できると報告されている15。この文献では、SEI成分を分析したところ、先の2014年の文献にある還元機構で説明でき、Li[FSA]が存在する場合としない場合で、リチウム金属とイオン液体の反応性が異なることを指摘している。2017年の論文では、イオン液体中におけるリチウム塩濃度が、リチウム金属の析出溶解挙動に及ぼす影響を調べたものがある16。この報告では、高濃度化(3.8 mol kg−1 Li[FSA]-[P111i4][FSA])することでSEIが安定化し、デンドライト成長が起こりにくくなるというものである。2020年の論文では、高濃度化(3.2 mol kg−1 Li[FSA]-[C3C1pyrr][FSA])することで、析出溶解時の分極が小さくなり、20 mA cm−2の高電流密度下での安定した析出溶解が可能になると報告されている17。
筆者らは温度という軸を増やすことでイオン液体の特性を生かすことを検討しており、2020年に室温から中温におけるイオン液体を用いたリチウム金属電池の性能を報告した18。Li[FSA]-[C2C1im][FSA]イオン液体を電解液とするビーカーセル中において、25 °Cで210分間リチウム金属の析出溶解を繰り返した(1サイクル8分間、8 mA cm−2)後のセルの様子を図5示す。上述の文献と同様に、有機溶媒の場合と比較して、イオン液体中では明らかにデンドライト生成が抑制されていることがわかる。また、コインセルを用いて25 °Cと90 °Cで分極の大きさやサイクル特性を比較すると、90 °Cのイオン液体中では分極が極めて小さく、安定な析出溶解サイクルが可能であることが分かった。Li/Li2FeP2O7ハーフセルでは高いレート特性とサイクル特性を実現している。
なおアニオンに注目が集まりがちであるが、有機カチオンの重要性も指摘されており、有機電解液に添加されたFSA−あるいはTFSA−イオン液体の有機カチオンがリチウム金属の平滑な析出溶解に有効であることが報告されている19(カチオンの機能性に注目して、イオン液体を金属電池用有機電解液の添加剤として利用するという同様の報告が近年よく見られる)。

図5 Li/Li対称セルで210分間析出溶解を行った後のリチウム金属の析出形態.8.0 mA cm−2, 25 °C, 8 min per cycle. IL: イオン液体電解質、OE: 有機電解液.イオン液体中ではデンドライト成長が抑制されていることがわかる。エルセビア社より許可を得て転載.18
4.ナトリウム金属電池におけるイオン液体
リチウム金属電池の場合と同様に、ナトリウム金属電池も高いエネルギー密度が魅力的なエネルギー貯蔵デバイスであるが、同様の問題点も有している20。イオン液体電解質はナトリウム金属電池への応用も検討されてきた21,22。なお、ナトリウム二次電池はリチウム二次電池と比較して、資源的制約が少なく、大型用途への展開が期待されている。ナトリウム二次電池の優位性については関連文献をご参照いただきたい23,24。
ナトリウム金属はリチウム金属より反応性が高く、イオン液体とも反応する。反応機構はリチウム金属の場合に似ていると考えられている。筆者らは2016年に、FSA−とTFSA−の影響(C2C1im+塩で比25)とNa塩の有無の影響に着目して、金属ナトリウムとイオン液体の反応性を調べた26。サイクリックボルタンメトリーの結果によると、FSA−イオン液体中ではカソードスキャンでナトリウム金属析出後、アノードスキャンでナトリウム金属の溶解が観測されるが、TFSA−イオン液体では溶解が観測されない。このことはアニオンによって、ナトリウム金属とイオン液体の反応性が異なることに由来すると考え、これを目視と紫外可視分光で調べた。ここでは変色したら反応が進んだと考えており、得られた結果を図6にまとめる。TFSA−のみを含むイオン液体(試料BとD)は、Na塩の有無に関わらず明確な着色が起こることがわかる。一方でFSA−のみを含むイオン液体(試料AとC)は、Na塩の有無に関わらずほとんど着色していないように見えるが、紫外可視分光ではNa塩を含まないもの(試料A)はわずかにスペクトルの変化が見える。興味深いのはFSA−とTFSA−が混合された系で、[C2C1im][TFSA]に10 %でもNa[FSA]塩が入ると着色せず(試料F)、1 %でもかなり着色は抑制される(試料G)。これらの結果は、FSA−イオン液体よりTFSA−イオン液体の方がナトリウム金属と反応しやすいこと、TFSA−イオン液体でもFSA−アニオンがあると反応しにくくなること、Na塩があると反応しにくくなることを示している。これらの結果はリチウム系での議論にも一致しており、FSA−の還元分解生成物が安定な被膜を形成することで説明できる。なお還元耐性の高いピロリジニウム系イオン液体では結果は異なる。
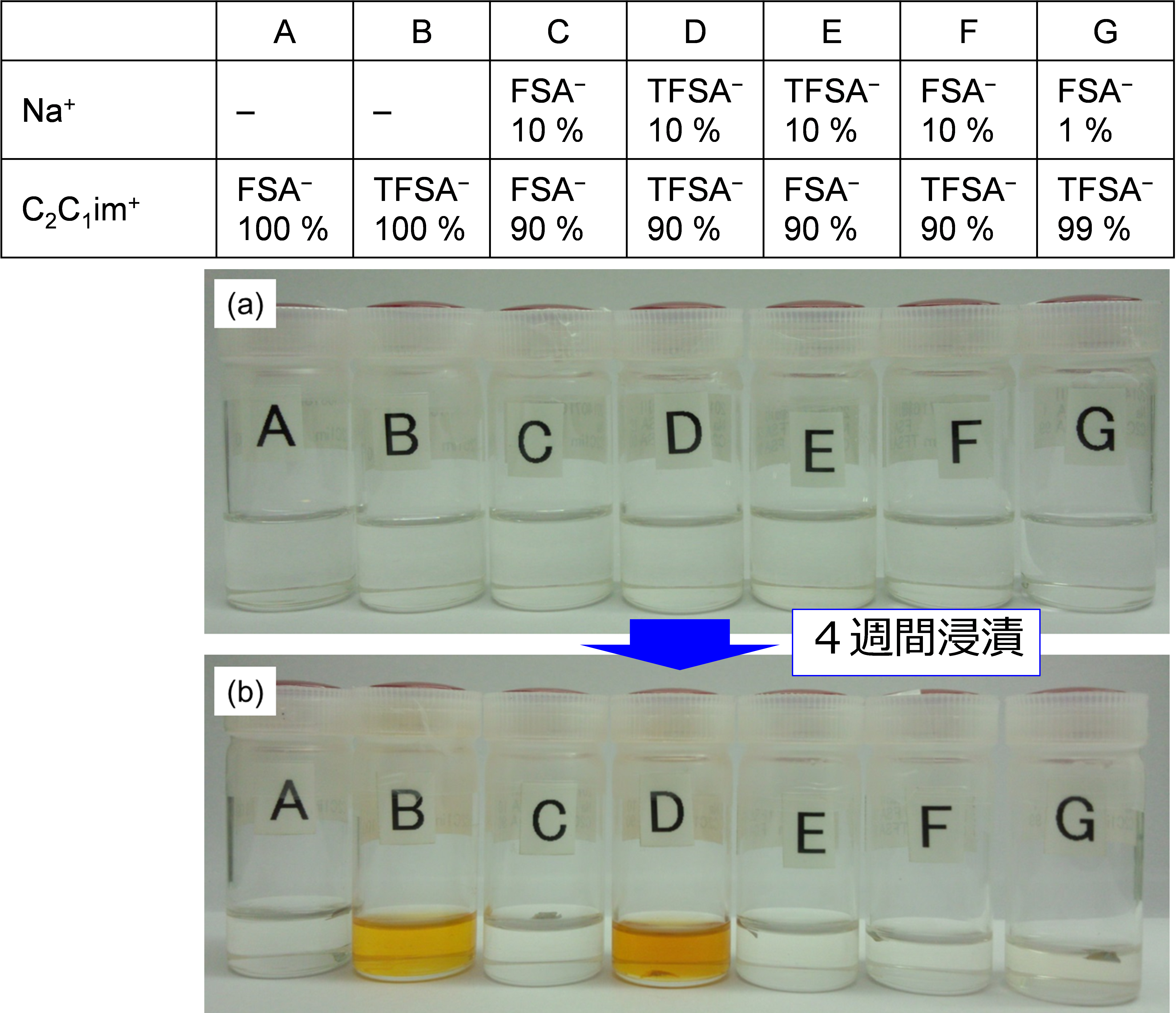
図6 様々なイオン液体中にナトリウム金属を4週間浸漬した後のイオン液体の様子.写真上部の表に混合した塩の割合がまとめられている.アメリカ化学会より許可を得て転載.26
Na[FSA]–[C2C1im][FSA]イオン液体中でのナトリウム金属の析出溶解挙動について、ビーカーセルを用いて、異なる温度で観察した結果を図7に示す27。0 °Cと25 °Cで観察されるデンドライト成長は90 °Cでは非常に起こりづらいことがわかる。この現象には温度が上がることでイオン液体中のNa+イオンの輸送が促進されること、融点近くで金属の表面拡散が促進されることなどが関わっていると考えている。この挙動は正極材料の長期試験をハーフセルで行う際に、対極のナトリウム金属の析出溶解が原因でサイクルができなくなるのを防ぐことに役立つことが分かっており、筆者らは5000サイクルを越えるハーフセル試験を実現している28。またこの性質を利用して、初期に負極に載せるナトリウム金属の量を制限したNa/Na3V2(PO4)3フルセル試験を実施したところ、負極と正極の容量比(N/P比)が2程度でも150サイクル以上作動できることを確認した29。初期に負極にナトリウム金属を載せないアノードフリー金属電池については、ハードルが極めて高い(図3)。筆者らはアルミニウム基板をアニーリング後フッ素化することで、結晶方位と表面形態・エネルギー(親ナトリウム性の高い基板)を制御できることを見出した30。このアルミニウム基板を負極集電体として、Na[FSA]-[C2C1im][FSA]イオン液体電解質と組み合わせて、アノードフリーナトリウム金属電池を作製した。25 °Cと90 °Cでサイクル特性を比較したところ、やはり90 °Cでは高い容量を維持できることが分かっており、集電体の前処理、イオン液体の利用、中温作動という組み合わせで、アノードフリー電池も高性能化できることを示した。

図7イオン液体中で析出したナトリウム金属の析出形態.左: 25 °C、右: 90 °C.0.2 mA cm−2, 0.2 C cm−2 per cycle.アメリカ電気化学会より許可を得て転載. 27
イオン液体の高い熱安定性を活かすと、液体ナトリウム金属電池の作動も可能である(ナトリウム金属の融点は97 °C)。NGKによって電力貯蔵用にナトリウム-硫黄(Na-S)電池が実用化されているが、Na-S電池は正極と負極をナトリウムイオン伝導性があるβ-アルミナ固体電解質(BASE)によって分離している(Na/BASE/S(NaxS)セル)31。正極室において生成する多硫化ナトリウム(NaxS)を溶融させるために300 °C以上で作動させることが求められるが、筆者らはイオン液体を正極室に導入すれば多硫化ナトリウムは固体でも問題ないと考え(Na/BASE/IL/S(NaxS)セル)、無機イオン液体を導入して150 °Cで作動するNa-Sを報告した(図8)32。このタイプの電池では、イオン液体はβ-アルミナ固体電解質によって液体ナトリウム金属と分離されていて直接触れないため、イオン液体の還元分解を考える必要はなく、イオン液体は正極室でナトリウムイオン伝導だけを担えばよい。残念ながら有機イオン液体はこの温度において、多硫化物イオンと反応するため使用できず、Na[TFSA]-Cs[TFSA]無機イオン液体を用いたが、1000サイクルを越える安定性を実現している。
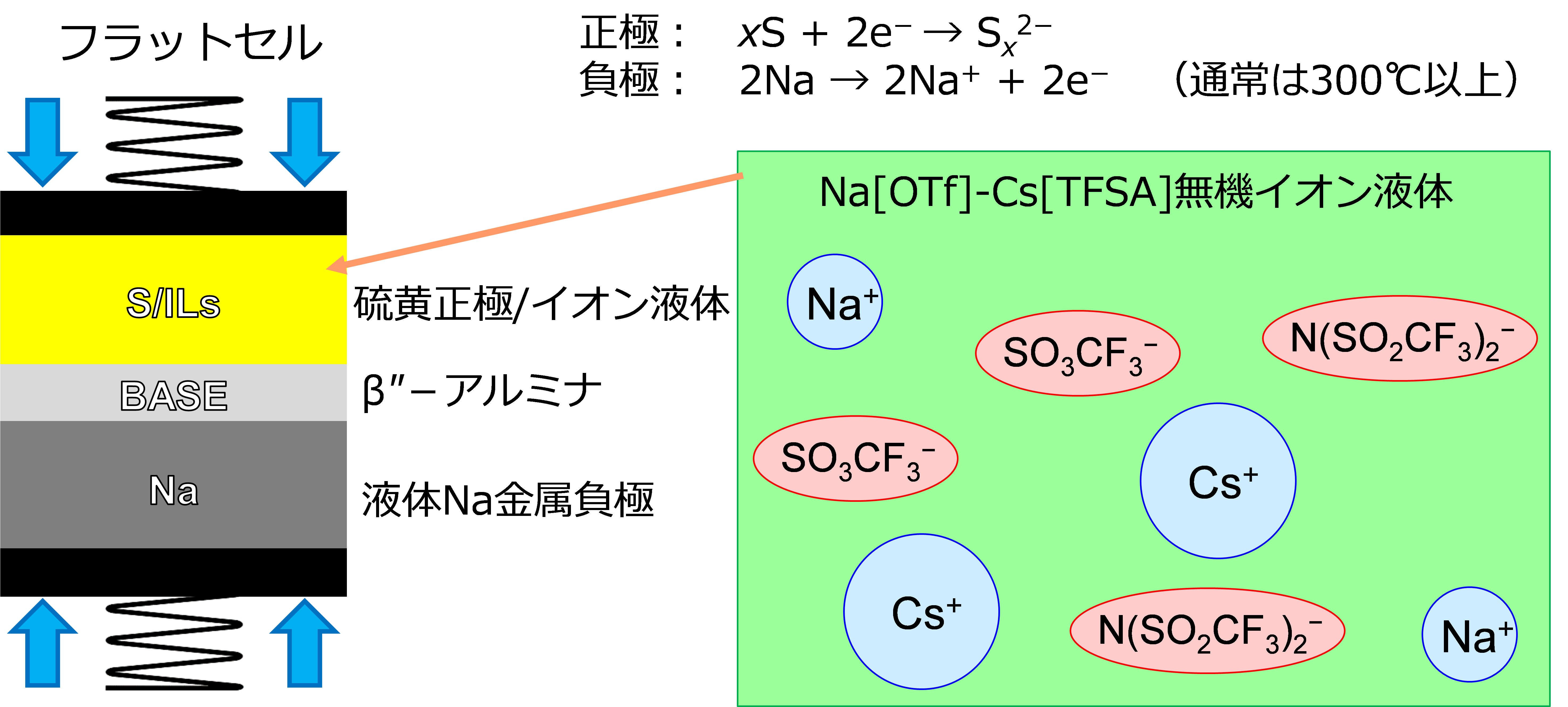
図8 無機イオン液体を用いた150 °Cで作動するNa-S電池の模式図.32
さらに温度を下げて、105 °Cで作動する液体ナトリウム金属電池にも取り組んだ。ここでは主に90 °Cで作動する固体ナトリウム金属電池との比較で、Na/BASE/IL/Na3V2(PO4)3セルの挙動を調べた(図9)33。正極室にイオン液体(Na[FSA]–[C2C1im][FSA])を導入しない場合、β-アルミナ固体電解質とNa3V2(PO4)3正極はいずれも固体であるため、イオン伝導が取れず電池の作動は難しい(一般的な固体電池と同じ問題)。ここにイオン液体をいれるとナトリウムイオン伝導性を与えることができ、電池として作動させることができる。このときナトリウム金属の融点以下では固体ナトリウム金属とβ-アルミナ固体電解質が固体同士であるため、やはりイオン伝導性が悪く、また析出溶解を繰り返すことで、デンドライト成長によるβ-アルミナ固体電解質の破損が起こる。つまり、液体ナトリウム金属の使用が有効であり、これを使える温度で安定にこのセルを作動させるには、正極室にイオン液体のような界面をつなぐ役割の電解質が必要である。実際にこの電池を作動させると、90°Cでは100サイクル程度で劣化してしまうが、150 °Cでは500サイクルでも劣化はほぼ起きない。

図9 (a) 固体ナトリウム金属と液体ナトリウム金属を用いたNa/BASE/IL/Na3V2(PO4)3セルの模式図.固体ナトリウム金属負極を用いたセルでサイクルした後のアルミナ固体電解質の(b)表面と(c)SEM像.液体ナトリウム金属負極の場合はこのような破損は観察されない.エルセビア社より許可を得て転載.33
5.おわりに本稿ではイオン液体を用いた金属電池について紹介した。現在実用化されている二次電池はイオン電池が主流だが、よりエネルギー密度の高い金属電池が実用化されれば、安くて長持ちな電池が実現するかもしれない。現実的な障壁はたくさんあるが、安全性の高いイオン液体電解質を用いることで少しでも実用化が近づけばよいと思う。あとイオン液体がもうちょっと安くなれば良いのだが、これは電池への応用に限ったことでもない。
参考文献
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda, K. Dokko, Chem. Rev. 117, 7190−7239 (2017).
- Electrochemical Aspects of Ionic Liquids (2nd ed.), Ed. H. Ohno, John Wiley & Sons, Inc., 2011, Hoboken, New Jersey.
- Y. Zheng, D. Wang, S. Kaushik, S. Zhang, T. Wada, J. Hwang, K. Matsumoto, R. Hagiwara, EnergyChem 4, 100075 (2022).
- T. Welton, Chem. Rev. 111, 3508–3576 (2011).
- R. Hagiwara, Y. Ito, J. Fluorine Chem. 105, 221-227 (2000).
- D. Wang, J. Qiu, N. Inui, R. Hagiwara, J. Hwang, K. Matsumoto, ACS Energy Lett. 8, 5248−5252 (2023).
- J.G. Zhang, W. Xu, J. Xiao, X. Cao, J. Liu, Chem. Rev. 120, 13312−13348 (2020).
- X.B. Cheng, R. Zhang, C.Z. Zhao, Q. Zhan, Chem. Rev. 117, 10403−10473 (2017).
- L. Yang, N.M. Hagh, J. Roy, E. Macciomei, J.R. Klein, U. Janakiraman, M. E. Fortier, J. Electrochem. Soc. 171 060504 (2024).
- H. Zhang, W. Feng, J., Z. Zhou, J. Fluorine Chem. 174, 49–61 (2015).
- H. Matsumoto, H. Sakaebe, K. Tatsumi, M. Kikuta, E. Ishiko, M. Kono, J. Power Sources 160, 1308–1313 (2006).
- M. Ishikawa, T. Sugimoto, M. Kikuta, E. Ishiko, M. Kono, J. Power Sources 162, 658–662 (2006).
- I.A. Shkrob, T.W. Marin, Y. Zhu, D.P. Abraham, J. Phys. Chem. C 118, 19661−19671 (2014).
- G.M.A. Girard, M. Hilder, N. Dupre, D. Guyomard, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D.R. MacFarlane, P.C. Howlett, ACS Appl. Mater. Interfaces 10, 6719−6729 (2018).
- A. Basile, A.I. Bhatt, A.P. O’Mullane, Nat. Commun. 7, 11794 (2016).
- G.M.A. Girard, M. Hilder, D. Nucciarone, K. Whitbread, S. Zavorine, M. Moser, M. Forsyth, D.R. MacFarlane, P.C. Howlett, J. Phys. Chem. C 121, 21087–21095 (2017).
- K. Periyapperuma, E. Arca, S. Harvey, C. Ban, A. Burrell, D.R. MacFarlane, C. Pozo-Gonzalo, M. Forsyth, P.C. Howlett, J. Mater. Chem. A 8, 3574–3579 (2020).
- J. Hwang, H. Okada, R. Haraguchi, S. Tawa, K. Matsumoto, R. Hagiwara, J. Power Sources 453, 227911 (2020).
- D.-J. Yoo, K.J. Kim, J.W. Choi, Adv. Energy Mater. 8, 1702744 (2018).
- B. Lee, E. Paek, D. Mitlin, S.W. Lee, Chem. Rev. 119, 5416−5460 (2019).
- K. Matsumoto, J. Hwang, S. Kaushik, C.Y. Chen, R. Hagiwara, Energy & Environ. Sci. 12, 3247–3287 (2019).
- A. Basile, M. Hilder, F. Makhlooghiazad, C. Pozo-Gonzalo, D. R. MacFarlane, P. C. Howlett, M. Forsyth, Adv. Energy Mater. 8, 1703491 (2018).
- J.M. Tarascon, Nature Chem. 2, 510 (2010)
- N. Yabuuchi, K. Kubota, M. Dahbi, S. Komaba, Chem. Rev. 114, 11636–11682 (2014).
- K. Matsumoto, T. Hosokawa, T. Nohira, R. Hagiwara, A. Fukunaga, K. Numata, E. Itani, S. Sakai, K. Nitta, S. Inazawa, J. Power Sources 265, 36–39 (2014).
- T. Hosokawa, K. Matsumoto, T. Nohira, R. Hagiwara, A. Fukunaga, S. Sakai, K. Nitta, J. Phys. Chem. C 120, 9628–9636 (2016).
- K. Matsumoto, C. Y. Chen, T. Kiko, J. Hwang, T. Hosokawa, T. Nohira, R. Hagiwara, ECS Trans. 75, 139–145 (2016).
- J. Hwang, K. Matsumoto, R. Hagiwara, Adv. Sustainable Syst. 2, 1700171(2018).
- S. Wu, T. Wada, H. Shionoya, J. Hwang, K. Matsumoto, R. Hagiwara, Energy Storage Materials 61, 102897 (2023).
- S. Wu, J. Hwang, K. Matsumoto, R. Hagiwara, Adv. Energy Mater. 13, 2302468 (2023).
- 玉越 富夫, 伊藤良幸, 電気化学, 91, 300–304 (2023).
- D. Wang, J. Hwang, C.Y. Chen, K. Kubota, K. Matsumoto, R. Hagiwara, Adv. Funct. Mater. 31 (2021) 2105524.
- J. Qiu, D. Wang, J. Hwang, K. Matsumoto, R. Hagiwara, J. Power Sources 612, 234777 (2024).