Topics
2022.03.31 circular No.18
イオン液体が拓くマグネシウム金属電池物質・材料研究機構 エネルギー・環境材料研究拠点 先進蓄電池研究開発拠点
万代 俊彦
凝り性なくせに堪え性がない。やり始めると細部まで徹底しないと気が済まない。にもかかわらず情熱を維持できない。キッチン収納には謎の調味料と謎のリキュールが所せましと並び、ただでさえ狭いキッチンをよりコンパクトな空間としていた。そんな自分が飽きもせず、6年以上も同じ研究テーマに挑戦している。どこにその魅力が、と問われても即答できない。そんな不思議な、好奇心をちょうど良い具合にかきたてる存在、それがマグネシウム金属電池(電解液)である。マグネシウム金属電池とは、負極活物質にマグネシウム金属を用いた蓄電池である。マグネシウムを電池利用する利点としては、主に材料コストと安全性が挙げられる。マグネシウムの地殻埋蔵量はリチウムの1700倍、海水からも採取できることから昨今懸念されている外交的なリスクが小さく、価格もアルミニウムに比肩する1。またリチウムをはじめとしたアルカリ金属とは異なり、マグネシウム金属は空気中で安定な酸化被膜を表面に形成するため安全に取り扱いが可能である。さらには2価の酸化還元反応を利用することができるため、グラム当たりの容量密度はリチウムイオン電池で用いられている炭素材料の約6倍にも上る(図1)。このようなマグネシウム金属の地質学的、化学的特長からマグネシウム金属電池は、コスト性・安全性・蓄電容量を備えた蓄電池として、例えば減災や防災に資する大容量エネルギー貯蔵デバイスへの展開が期待されている。
電池は大きく分けて正極・負極・電解質の3要素から構成されており、容量や出力など電池の理論性能を直接決定づけるのは、正極および負極である(言い換えれば容量や出力の計算に電解質は関与しない)。しかし実電池において、電池性能は電解質の種類に強く依存する。電解質は、正極と負極を物理的に隔て、キャリアイオンの授受および輸送をもって正負極間における電極反応を仲介する。すなわち、電極と電解質の界面におけるキャリアイオンの授受および電解質中のイオン輸送のしやすさが電極反応に多大な影響を与え、電池の実効性能が変化する。そのため既に商用化されているリチウムイオン電池でさえも、その性能を最大限引き出すために、より良い電解質を見つけ出すべく多くの企業で研究開発が精力的に行われている。MgMn2O4正極を例として、マグネシウム金属電池の電池式は下記のように表される(係数は省略)。
(正極) MgMn2O4 + Mg2+ + 2e− ⇄ Mg2Mn2O4
(負極) Mg ⇄ Mg2+ + 2e−
(全反応) MgMn2O4 + Mg ⇄ Mg2Mn2O4
マグネシウム金属の電気化学的溶解析出反応が、負極の基礎反応であることがお分かりいただけるかと思う。式で表すと非常に簡単なこの負極反応こそが、マグネシウム金属電池実現における最大の障壁なのである。本稿ではまず、マグネシウム金属電池電解質に求められる要件を整理したい。厳しい現実が待っている。しかし厳しくともどこかに活路はあると信じ研究を続けた結果、幸運にも筆者はそれを馴染みのあるイオン液体に見出した。イオン液体の優しさ(包容力)が染みた。残念ながら現在は非イオン液体の電解液系にシフトしつつあるが、イオン液体電解質はマグネシウム金属負極の学理の深化に著しく貢献し、マグネシウム金属電池研究ステージを躍進させた立役者であることに疑いの余地はない。ここではイオン液体が拓いたマグネシウム金属電池のサイエンスについて、具体例を交えながら解説する。
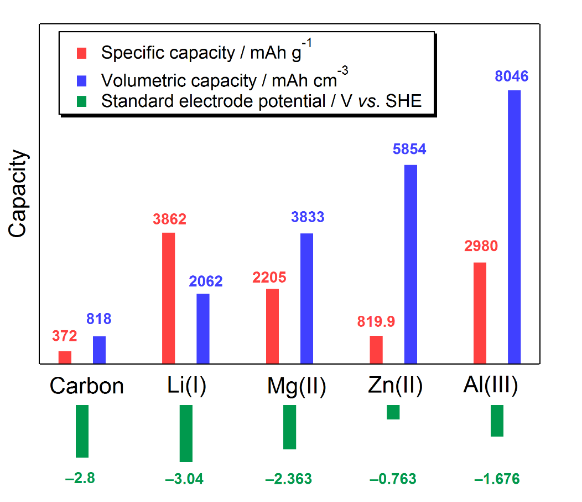
図1. 各種負極活物質の容量密度および電極電位
2.マグネシウム金属電池電解質開発の難しさ電解質への要件はいくつか挙げられるが、中でも『マグネシウム析出溶解活性を有すること』が難しく、電解質構成要素の選択肢がかなり絞られる。図2に筆者がこれまで検討してきた成果に文献情報を加えてまとめた、電解液組成とマグネシウム析出溶解活性の関係図を示す(busyな図で申し訳なく思う)。矢印の多くが不活性・低活性に向かっている。まさに死屍累々である。溶媒の選択肢としては、エーテルと一部のジアルキルスルホンに限定される2。マグネシウム塩の対アニオンに関して、イオン液体でも馴染みのあるBF4−, PF6− , CF3SO3−はマグネシウム金属との親和性が低く、電解液成分として好ましくない3–7。近年電池分野でホットなbis(fluorosulfonyl)amide (FSA)に至っては、マグネシウム金属をほぼ完全に不活化してしまう8,9 。その点リチウム金属はニトリルやアミドなど含窒素溶媒とは相性が悪いが、多種多様な溶媒・アニオンを許容でき、包容力に圧倒的な差がある。マグネシウム系電解質の多くは、支持塩+溶媒から成る電解質にさらにハロゲン化物を添加すると活性が発現あるいは向上する(図2参照)。活性発現/向上の原理については後述するが、ハロゲン化物の添加は同時に腐食問題をもたらす。電池は構成材の多くが金属であるため、電解質は非腐食性である必要がある。このようにマグネシウム電池電解液開発においては、極めて限定された探索領域からマグネシウム塩および溶媒を選び出さなければならず、幅広い知見に加え、折れない精神力・忍耐力・柔軟性が試される。
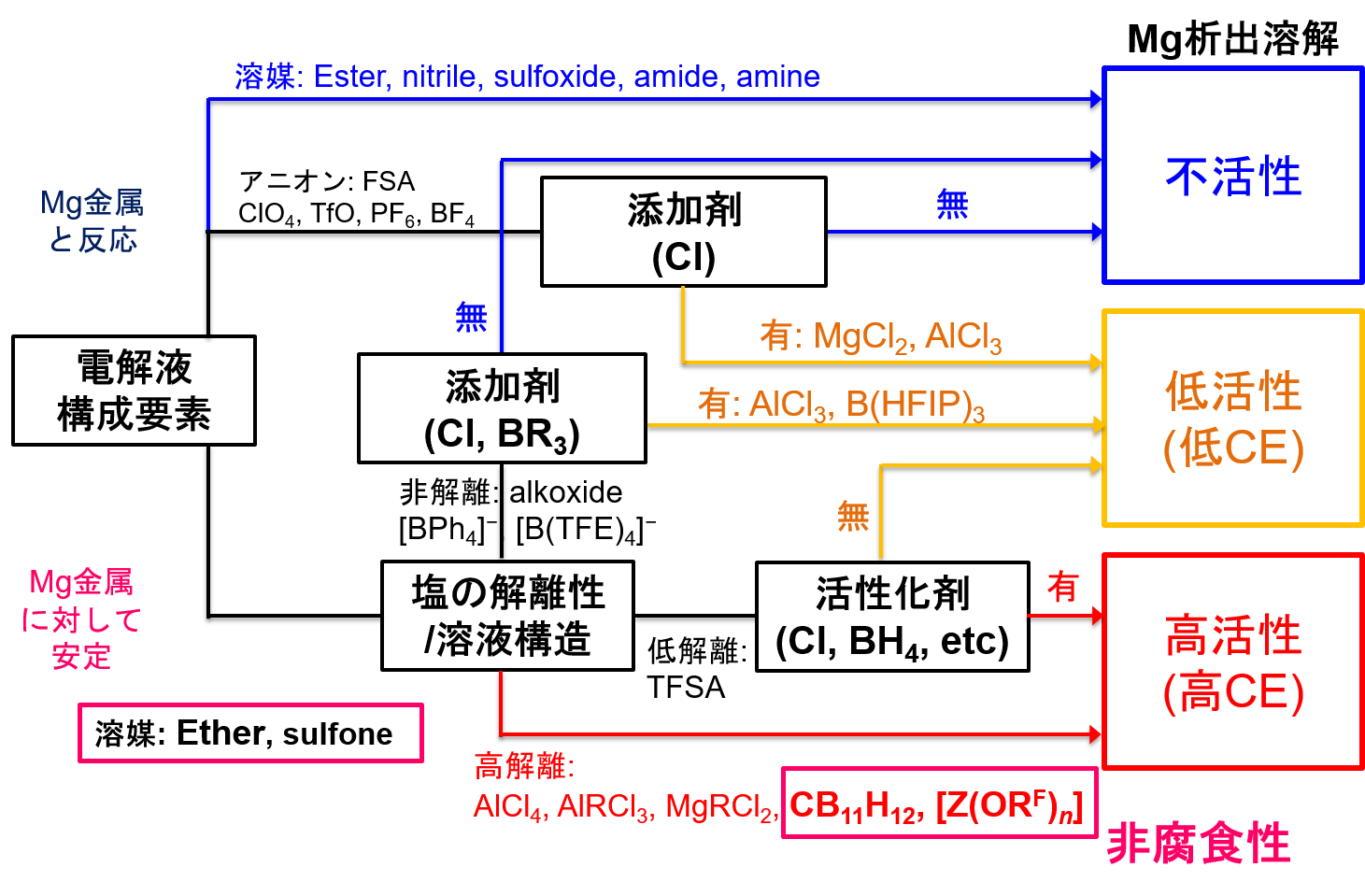
図2. 電解液構成要素とマグネシウム析出溶解活性の関係図
3.イオン液体×マグネシウム金属電池3.1 補助溶媒としてのイオン液体
そんな気難しいマグネシウム電解質にあって、イオン液体とは比較的良い関係を構築している(ように思う)。次節で少し説明するが、bis(trifluoromethanesulfonyl)amide (TFSA)系イオン液体はマグネシウム金属との親和性に優れる。そのためテトラヒドロフランなどの低沸点エーテルを主成分とする典型的なハロゲン系マグネシウム電解質と[TFSA]系イオン液体を混合することで、マグネシウム析出溶解活性を維持しながら耐熱性の大幅な向上が見込める。詳細は森田らの包括的な検討を参照されたい10-12。Mg[TFSA]2をグライム(glyme; Gn = CH3O(CH2CH2O)nCH3 )に代表されるオリゴエーテルに溶解した電解液は、マグネシウム析出溶解活性を示す数少ない非腐食性電解液である13-16。北田らはMg[TFSA]2 /glyme/イオン液体で構成される電解液からのマグネシウム電析を報告している17,18。汎用マグネシウム電解液へのイオン液体混合は、耐熱性もさることながらイオン伝導度の向上にも寄与しており、マグネシウム電析にかかる電流密度が1桁程度増大する効果も認められている。
3.2 主溶媒としてのイオン液体3.2.1 イオン液体とマグネシウム金属の親和性
エーテル-イオン液体混合溶媒は電解液特性改善に一定の効果はあるものの、エーテルの揮発性は依然課題として残る。高エネルギー密度のマグネシウム金属電池を実現するには遷移金属酸化物を正極活物質とする必要があり19、酸化物の結晶格子中でマグネシウムイオンを円滑に拡散させるためには高温条件が必須となる20。また高温では遷移金属イオンのアノード溶解が促進されるため、それを抑制する機能も求められる(実はこの機能こそがイオン液体をマグネシウム電解液に適用した最たる動機である)。特定物質(イオン)の溶解抑制と聞いて想起されるのが、イオン液体の異常溶解特性である。渡邉らはカチオン、アニオンともに弱配位性のイオンのみで構成されるイオン液体が、リチウム硫黄電池における往年の課題であった多硫化物(ポリスルフィド)の溶解を抑制するキーマテリアルであることを見出し、液体電解質を用いたリチウム硫黄電池のコンセプトを実証するに至った21–24。耐熱性、不揮発性、さらに異常溶解特性を備えたイオン液体は、マグネシウム金属電池電解質としてこれ以上ない適切な溶媒と言える。[TFSA]−を始め、アミドアニオンを対アニオンとしたイオン液体は熱安定性に優れる。そこでまず、これらアミドアニオン系イオン液体とマグネシウム金属の親和性を系統的に評価した。カチオンには電気化学的な安定性に優れる脂環式のN-methyl-N-propylpyrrolidinium [PYR13] +を選択した。マグネシウム片を各種イオン液体に25 °Cあるいは100 °Cで24h浸漬し,洗浄・乾燥後の表面の化学組成をエネルギー分散型X線分析(EDX)およびX線光電子分光法(XPS)により分析した。EDX分析結果を図3に示す。明確なアニオン依存性が認められ、[PYR13][TFSA]に浸漬したマグネシウム片ではMg元素比率が高い水準を維持していたのに対し、bis(pentafluoroethanesulfonyl)amide[BETA]−や[FSA]−、dicyanoamide [DCA]−に浸漬したマグネシウム片にはマグネシウム以外の元素(FやS、N)が多量に検出された。ページ数の都合で詳細は省くが、[BETA]−、[FSA]−、[DCA]−それぞれ還元的に弱い結合があるらしく、還元剤であるマグネシウムにより結合が切れ、マグネシウム表面に分解物が堆積することがXPSスペクトルから分かった([FSA]−、[DCA]−については大方の予想がついていたが、化学的性質が[TFSA]−に類似した[BETA]−も分解してしまう点は興味深い)。[PYR13][TFSA]は、マグネシウム電析を阻害しないこともGrignard試薬を含む混合電解液からの電析試験から分かっており、マグネシウム電解液の溶媒として最適なイオン液体の一つであることが伺える(詳細は拙著参照9)。
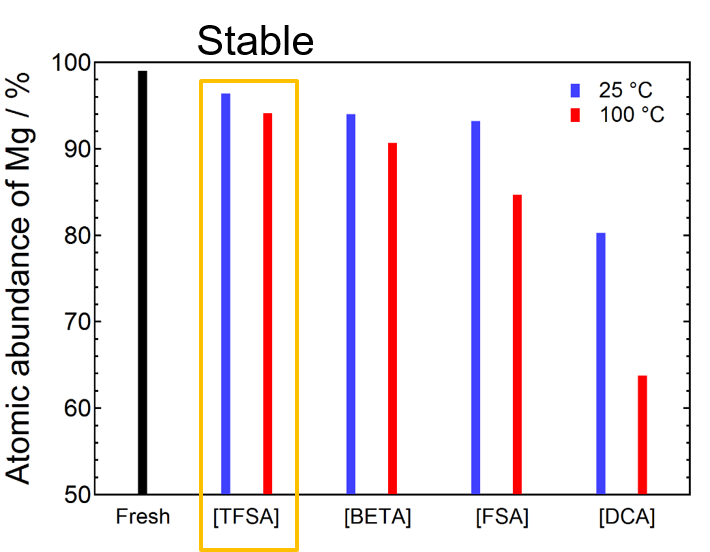
図3. [PYR13][amide]に浸漬したマグネシウム片表面におけるマグネシウム元素存在比率。
3.2.2 溶媒和構造制御: 反応の原理解明と電解液設計による副反応抑制以上の予備検討を踏まえて、[PYR13][TFSA]を溶媒に、Mg[TFSA]2 を支持塩とした電解液で電気化学的マグネシウム析出溶解が可能かと言うと、そうは問屋が卸さないのがマグネシウム金属負極のつれないところである。(ごく一部例外はあるものの)イオン液体+マグネシウム塩からのマグネシウム電析は極めて困難である。Mg(II)は強ルイス酸であるためルイス塩基であるアニオンを強く引き付ける。例えばMg[TFSA]2/[PYR14][TFSA]中には [Mg(TFSA)3]−や[Mg(TFSA)4]2−が、Mg[TFSA]2/[PYR1,2O1][TFSA]から構成される結晶にはn[Mg(TFSA)3]n−というポリアニオンが存在することが示されている25,26。マグネシウムを電気化学的に析出させるためにはMg(II)からアニオンを引きはがす必要があるが、2価の正電荷からアニオンを取り払うのは困難であろう。そこで高解離度を期待してMg[B(HFIP)4]2(HFIP = hexafluoro-iso-propoxyl)を支持塩として[PYR13][TFSA]に溶解させてみたところ、残念なことに[PYR13]2[TFSA][B(HFIP)4]で表されるイオン性固体が即座に析出した...(図4。忍耐力が試された場面の一つ)。見方を変えて活性な電解液に目を向ける。前述のように、Mg[TFSA]2/glymeは電気化学的マグネシウム析出溶解活性を発現する。つまり溶媒であるglymeが活性を決定づける鍵であることが推察される。Mg[TFSA]2とglymeは、glyme鎖長や組成比によって様々な溶媒和物を形成する。例えばMg[TFSA]2とtetraglyme (G4)はモル比1:1で安定錯体を形成する16,18。この錯体は融点が約140°Cであり、融解後も220 °C程度まではその構造が維持される16。錯体の融液を電解液としたところ(広義の溶媒和イオン液体)、予想通りマグネシウム析出溶解活性を発現した(図5)。このことからマグネシウム-エーテル溶媒和物が、電気化学活性種であることが分かる。そこでMg[TFSA]2ではなく、[Mg(G4)][TFSA]2を支持塩として改めて[PYR13][TFSA]に溶解した電解液について電気化学試験を実施すると、マグネシウム析出溶解活性を示した(図5)9。溶液のラマン分光分析および溶液から析出した固体の結晶構造解析から、溶媒和構造[Mg(G4)]が溶液中でも保持されていることが示唆され、これらの結果もマグネシウム-エーテル溶媒和物が活性種であることを支持している。
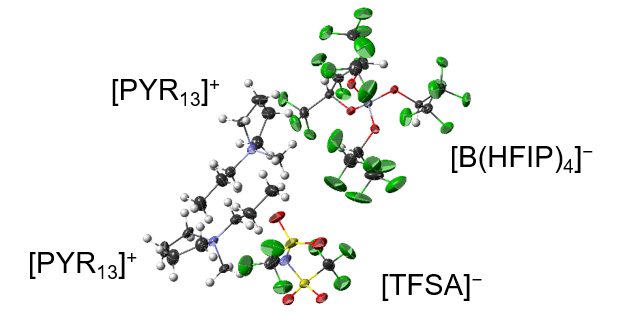
図4. [PYR13]2[TFSA][B(HFIP)4]の結晶構造。Mg[B(HFIP)4]2/[PYR13][TFSA]から析出。
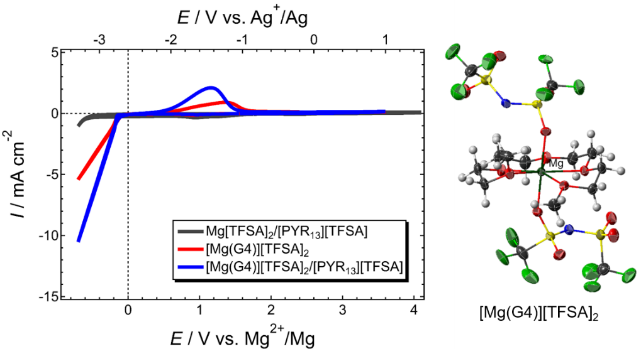
図5. (左)[Mg(G4)][TFSA]2融液および[Mg(G4)][TFSA]2/[PYR13][TFSA]中におけるPt電極のサイクリックボルタモグラム。Mg[TFSA]2/[PYR13][TFSA]の測定結果も併せて掲載。(右)[Mg(G4)][TFSA] 2の結晶構造。
さて、晴れて電気化学的マグネシウム析出溶解活性なイオン液体電解液が誕生したわけだが、析出溶解の効率は20%程度であり、析出反応の大部分を副反応が占めていることが分かる。電析物の元素分析の結果、[Mg(G4)][TFSA]2/[PYR13][TFSA]電解液では[TFSA]− の還元分解が析出反応と同時に進行していることが判明した。他方、同じMg塩を支持塩とするMg[TFSA]2/glymeにハロゲン化物を添加すると活性および効率が劇的に改善し、高純度のマグネシウム析出すら可能である27–29 。ハロゲン化物添加は[TFSA]−分解を抑制する効果がある。それでは[TFSA]−の還元耐性を決定づける物理化学的な因子は何だろうか。答えは[TFSA]−の配位状態にあった。前述したように、イオン液体電解液中において [Mg(G4)][TFSA]2 構造は維持されている。すなわち、[TFSA]−はMg(II)に対して直接配位しているのである(図5)。一方、ハロゲン化物含有電解液ではハロゲン化物による配位交換が起こり、[TFSA]−が解離している27–29。 [TFSA]−の解離状態と還元安定性は量子化学計算によりその関係性が示されており9,30、端的に言うとMg(II)に配位した[TFSA]−はMg (II)の電場により大きく分極し、還元耐性が著しく低下する(図6)。言い換えれば[TFSA]−を非腐食性かつ耐酸化性に優れる配位子により解離させることができれば、活性のみ向上させることも可能となる。そこで[Mg(G4)][TFSA]2に2当量の適当な分子を加え、溶媒和構造の制御を試みた。図7に[Mg(G4)(DPSO2) 2][TFSA]2および[Mg(G4)(MIm)2][TFSA]2 の結晶構造を示す(DPSO2: dipropylsulfone, MIm: methylimidazole)。結晶中で[TFSA]−に替わりこれらの有機分子がMg(II)に直接配位しており、狙い通り[TFSA]−を解離させることができた。尚、過剰量のMImを添加すると[Mg(MIm)6][TFSA]2が生成することから、『2当量』というのが重要なのであろう。これらの新規溶媒和物を支持塩としたイオン液体電解液の電解液特性を評価した結果、配位子依存性が確認された(図8)。[Mg(G4)(MIm)2][TFSA] 2 の場合は還元電流のみが観測され、対応する酸化電流は一切観測されなかった。[Mg(MIm)6][TFSA]2 がマグネシウム析出溶解不活性であることから、MImにより電析Mgが即座に不活化されることが考えられる。一方[Mg(G4)(EPSO 2)2][TFSA]2を支持塩とするとマグネシウム析出溶解活性が著しく向上した。析出溶解効率は50%超にも達した。電析物に含まれる[TFSA]−由来の成分が著しく減少していることから、[TFSA]− をMg(II)から完全に解離させることが電解質の耐還元性を高めることに有効であることが実証された9。
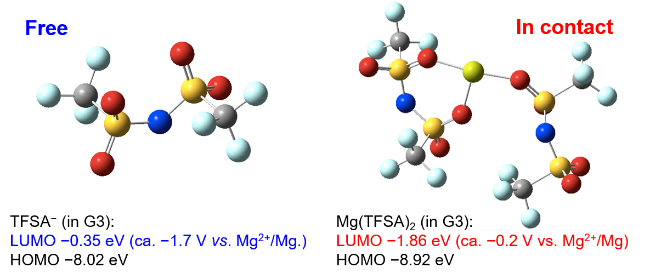
図6. [TFSA]の配位状態によるHOMO-LUMO準位の違い。
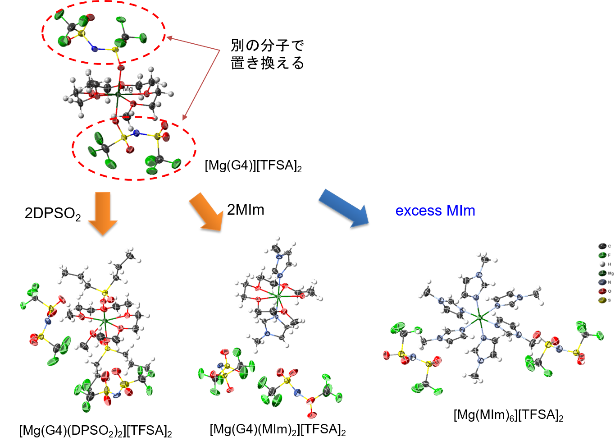
図7. 各種Mg錯体の結晶構造
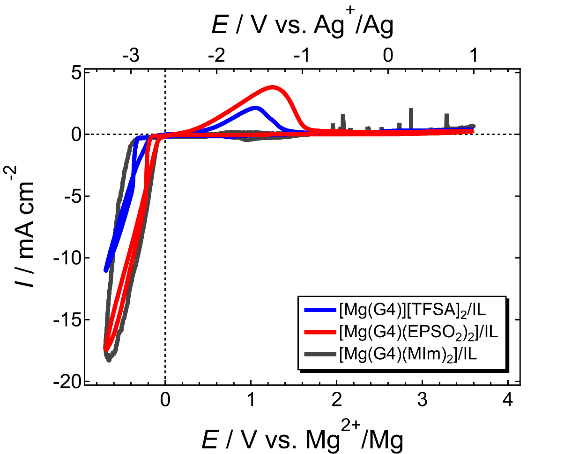
図8. [Mg(G4)(L)2][TFSA]2/[PYR13][TFSA]の電気化学特性
イオン液体の”デザイナー性”を利用することで、分子性溶媒フリーなイオン液体電解質も創製できる。本質的なコンセプトはMg錯体/イオン液体電解液と同じである。ピロリジニウムカチオンや4級アンモニウムカチオンにエーテル結合(オリゴエーテル)を導入することで側鎖にエーテル性配位子能を付与、さらにMg(BH4)2を支持塩とすることで、イオン液体電解液中でもマグネシウムの電気化学的析出溶解が可能となる(BH4−は[TFSA]−よりもルイス塩基性が強く、またMg(II)に直接配位してもマグネシウムの酸化還元電位で分解されない耐還元性を持つ)8,31。ただオリゴエーテル基の導入は粘性の大幅な上昇を招くため、円滑なイオン輸送が求められる電解質においては不利である。さらにBH4−は耐酸化性に乏しく電池の高電圧化が困難なこともあって、カチオンのみならずアニオンの設計に対してもさらに深く踏み込む必要がある。
3.3 無機(中温)イオン液体(溶融塩)無機塩のみから構成される無機イオン液体(溶融塩)からもマグネシウムの電気化学的析出溶解が可能である。例えばMg[TFSA]2とCs[TFSA]からなる中温イオン液体(融点 > 100°C)はそれ単独ではマグネシウム析出溶解不活性であるが、Li[TFSA]を少量加えると活性を発現する32,33。この無機イオン液体は150°C付近で安定な液体として取り扱いができ、さらに耐酸化性に優れる。室温で固体であるが故の電池作製におけるテクニカルな面に目をつぶれば、耐熱性・耐酸化性が強く求められるマグネシウム金属電池に適した電解液の一つである。
3.4 電池適用: イオン液体が拓く3 V級マグネシウム金属電池最後にイオン液体系電解質の電池適用性について述べる。高電位作動正極の一つであるスピネル構造の酸化物を安定に充放電するためには3.7 V vs. Mg2+/Mg以上の耐酸化性(Eox)が求められており、一般的なエーテル系電解液(Eox = 3.3 V vs. Mg2+/Mg)は充電過程で分解してしまう。さらに遷移金属イオンのアノード溶解も競合するため、充電反応は満足に進まない。一方、Mg錯体/[PYR13][TFSA]の酸化端(Eox)は4.1 Vvs. Mg2+ /Mgであり、世界最高水準の耐酸化性を誇る。イオン液体を溶媒とすることでアノード溶解も劇的に抑制でき、MgCo2O4正極の安定な充放電を達成した(図9)。支持塩を[Mg(G4)(EPSO2)2][TFSA]2とすることで、充放電特性はさらに向上した(図10)。アニオンを完全に解離させたことによりMg(II)のイオン輸送特性や活量が上昇したことが、優れた充放電特性の要因と考えられる。
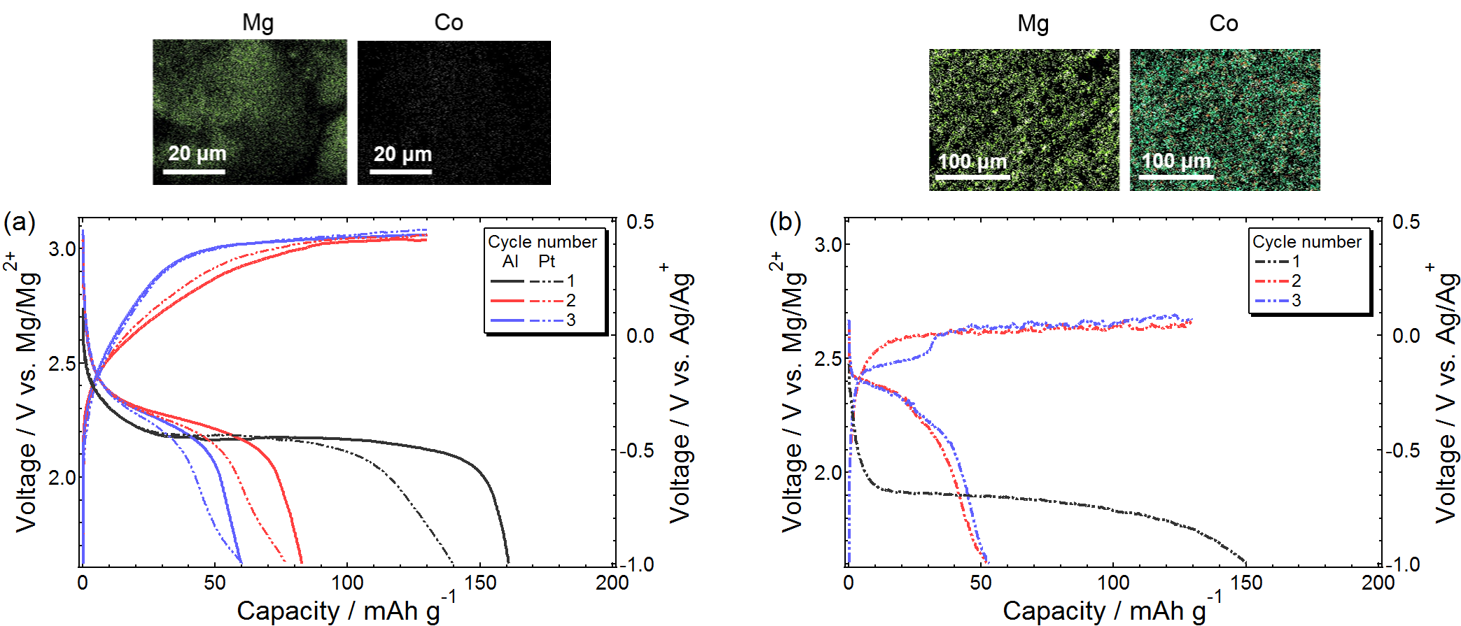
図9. (a) [Mg(G4)][TFSA]2/[PYR13][TFSA]、(b) Mg[TFSA]2/(G4+[PYR13][TFSA] (1:1 v/v))中におけるMgCo2O4正極の充放電挙動および、サイクル後のMg負極表面のEDX分析結果。 Reproduced from Ref. 9 with permission from the PCCP Owner Societies.
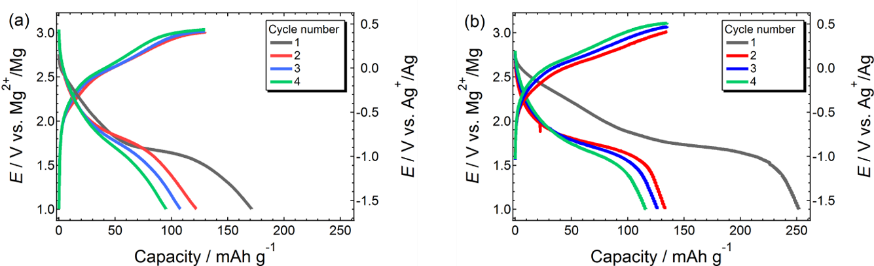
図10. MgMn2O4 正極の充放電特性。電解液の支持塩として、(a) [Mg(G4)][TFSA]2、(b)[Mg(G4)(EPSO2)2][TFSA]2を使用。 Reproduced from Ref. 9 with permission from the PCCP Owner Societies.
Mg錯体/[PYR13][TFSA]電解液は室温から200 °Cまで安定な液体として取り扱いできるため、特殊なセットアップや専門技術がなくとも簡単に電池を組むことができる。さらに耐酸化性のおかげで、これまで充電することすらできなかった正極の充放電ができるようになり、正極材料の研究開発が著しく活発化した。特に正極特性を最大限引き出す微細構造の理解が劇的に進展し、電池電圧約3Vのマグネシウム金属電池の動作実証に至った。繰り返しになるが、耐熱性・耐酸化性・異常溶解特性のどれが欠けてもこれを達成することは不可能に近く、まさにイオン液体だからこそ切り拓けた地平だと強く思う。
4. おわりに
本稿では、マグネシウム金属電池の特に電解液にフォーカスし、筆者が直面していた開発課題を述べるとともに、イオン液体を武器に切り拓いてきたマグネシウム負極・電解液のサイエンスを紹介した。イオン液体電解液の恩恵はここで述べたものに留まらず、特に正極開発は飛躍的に進展し、ついには室温でも円滑にMg2+の挿入脱離が可能な活物質も見出されてきている34,35。作動温度低下に伴い電解液もイオン液体系から弱配位性アニオンを用いたエーテル溶媒系にシフトしつつあるが36,37、室温作動の3V級マグネシウム金属電池実現も夢ではなくなってきた(図11)。マグネシウム金属電池は次々世代電池の一つであり、実現にはこの先も沢山のブレイクスルー、パラダイムシフトが求められる。最終的な電池の構成は想像もできないが、失敗と成功ひっくるめたこれまでの研究のすべてが実電池化の礎となると信じて、引き続き情熱を持って取り組んでいきたい。
思えば人生の岐路にはイオン液体があった。何を隠そう、妻との出会いはイオン液体討論会の懇親会である。新型コロナウィルスの感染拡大が、そうした男女の出会いの場を奪っているというのは由々しき事態である。一日でも早く落ち着き、対面での研究会および討論会が再開することを心から願う。イオン液体がもたらした個人的な”えん”はさらに続き、千葉大学西川恵子教授の下で学位取得後は横浜国立大学・渡邊正義教授の研究室に産学連携研究員として採用いただいた。渡邊教授にはさらに海外留学も後押しいただいた。帰国後、マグネシウム金属電池分野での躍進を支えてくれたのもイオン液体であり、私の人生を形作ってきたのはイオン液体と言っても過言ではない。などと調子いいことを言いながら、ここ最近は年間通して20時間程度しかイオン液体と戯れておらず、つかず離れず(若干離れ気味)の距離感に留まっている。そこは長距離相互作用を持つイオン液体である。多少離れても戻ってきたいと思わせる魅力がある。今後も適度な距離感で末永いお付き合いを続けていければと、イオン液体への一方的な慕情を吐露したところで本稿を締めたい。
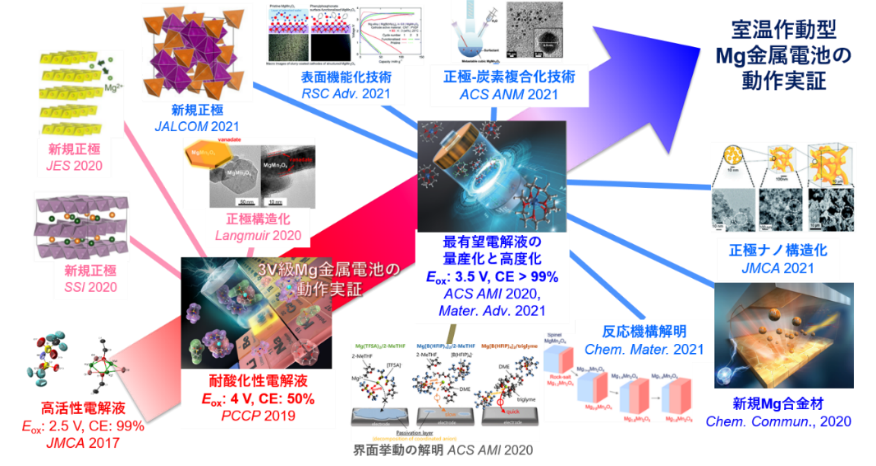
図11. 主要な研究成果とそれによってもたらされた当該分野の発展(Eox: 耐酸化性, CE: 負極反応効率)
謝辞本稿で取り上げた筆者の研究成果は、筆者が首都大学東京(現東京都立大学)、岩手大学、物質・材料研究機構在籍中に得られたものである。これらの研究は先端的低炭素化技術開発-次世代蓄電池(JST LCA-SPRING; Grant No. JPMJAL1301)および先進蓄電池研究開発拠点(JST COI-NEXT; Grant No. JPMJPF1601)の支援を受けて実施された。関係各位に謝意を表す。
参考文献
- Mineral commodity annual report 2019, https://www.mineralcommodities.com/wp-content/uploads/2020/04/LCM752_Annual-Report-2019_vFs.pdf
- S.-J. Kang, S.-C. Lim, H. Kim, J. W. Heo, S. Hwang, M. Jang, D. Yang, S.-T. Hong, H. Lee, Chem. Mater., 2017, 29, 3174–3180.
- G. Vardar, A. E. S. Sleightholme, J. Naruse, H. Hiramatsu, D. J. Siegel, C. W. Monroe, ACS Appl. Mater. Interfaces, 2014, 6, 18033–18039
- Z. Zhang, Z. Cui, L. Qiao, J. Guan, H. Xu, X. Wang, P. Hu, H. Du, S. Li, X. Zhou, S. Dong, Z. Liu, G. Cui, L. Chen, Adv. Energy Mater., 2017, 1602055
- E. N. Keyzer, H. F. J. Glass, Z. Liu, P. M. Bayley, S. E. Dutton, C. P. Grey, D. S. Wright, J. Am. Chem. Soc., 2016, 138, 8682–8685
- I. Shterenberg, M. Salama, Y. Gofer, D. Aurbach, Langmuir, 2017, 33, 9472–9478
- Y. Yang, W. Wang, Y. Nuli, J. Yang, J. Wang, ACS Appl. Mater. Interfaces, 2019, 11, 9062–9072.
- M. Kar, Z. Ma, L. M. Azofra, K. Chen, M. Forsyth, D. R. MacFarlane, Chem. Commun., 2016, 52, 4033–4036
- T. Mandai, K. Tatesaka, K. Soh, H. Masu, A. Choudhary, Y. Tateyama, R. Ise, H. Imai, T. Takeguchi, K. Kanamura, Phys. Chem. Chem. Phys., 2020, 21, 12100–12111
- 吉本信子, 森田昌行, Electrochemistry, 2012, 80, 104–108.
- T. Kakibe, N. Yoshimoto, M. Egashira, M. Morita, Electrochem. Commun., 2010, 12, 1630–1633.
- N. Yoshimoto, M. Matsumoto, M. Egashira, M. Morita, J. Power Sources, 2010, 195, 2096–2098.
- T. Fukutsuka, K. Asaka, A. Inoo, R. Yasui, K. Miyazaki, T. Abe, K. Nishio, Y. Uchimoto, Chem. Lett., 2014, 43, 1788–1790.
- Y. Orikasa, T. Masese, Y. Koyama, T. Mori, M. Hattori, K. Yamamoto, T. Okado, Z.-D. Huang, T. Minato, C. Tassel, J. Kim, Y. Kobayashi, T. Abe, H. Kageyama, Y. Uchimoto, Sci. Rep., 2014, 4, 5622.
- S. Y. Ha, Y.-W. Lee, S. W. Woo, B. Koo, J. S. Kim, J. Cho, K. T. Lee, N.-S. Choi, ACS Appl. Mater. Interfaces, 2014, 6, 4063–4073
- S. Terada, T. Mandai, S. Suzuki, S. Tsuzuki, K. Watanabe, Y. Kamei, K. Ueno, K. Dokko and M. Watanabe, J. Phys. Chem. C, 2016, 120, 1353–1365.
- A. Kitada, Y. Kang, Y. Uchimoto, K. Murase, J. Electrochem. Soc., 2014, 161, D102–D106.
- A. Kitada, Y. Kang, K. Matsumoto, K. Fukami, R. Hagiwara, K. Murase, J. Electrochem. Soc., 2015, 162, D389–D396.
- I. D. Johnson, B. J. Ingram, J. Cabana, ACS Energy Lett., 2021, 6, 1892–1900.
- M. Liu, Z. Rong, R. Malik, P. Canepa, A. Jain, G. Ceder, K. A. Persson, Energy Environ. Sci., 2015, 8, 964–974.
- J.-W. Park, K. Yamauchi, E. Takashima, N. Tachikawa, K. Ueno, K. Dokko, M. Watanabe, J. Phys. Chem. C, 2013, 117, 4431–4440.
- K. Ueno, J.-W. Park, A. Yamazaki, T. Mandai, N. Tachikawa, K. Dokko, M. Watanabe, J. Phys. Chem. C, 2013, 117, 20509–20516.
- K. Dokko, N. Tachikawa, K. Yamauchi, M. Tsuchiya, A. Yamazaki, E. Takashima, J.-W. Park, K. Ueno, S. Seki, N. Serizawa, M. Watanabe, J. Electrochem. Soc., 2013, 160, A1304–A1310.
- H. Moon, R. Tatara, T. Mandai, K. Ueno, K. Yoshida, N. Tachikawa, T. Yasuda, K. Dokko, M. Watanabe, J. Phys. Chem. C, 2014, 118, 20246–20256.
- G. A. Giffin, A. Moretti, S. Jeong, S. Passerini, J. Phys. Chem. C, 2014, 118, 9966–9973.
- G. A. Giffin, J. Tannert, S. Jeong, W. Uhl, S. Passerini, J. Phys. Chem. C, 2015, 119, 5878–5887.
- M. Salama, I. Shterenberg, L. J. W. Shimon, K. K. Adamsky, M. Afri, Y. Gofer, D. Aurbach, J. Phys. Chem. C, 2017, 121, 24909–24918.
- Y. Cheng, R. M. Stolley, K. S. Han, Y. Shao, B. W. Arey, N. M. Washton, K. T. Mueller, M. L. Helm, V. L. Sprenkle, J. Liu, G. Li, Phys. Chem. Chem. Phys., 2015, 17, 13307–13314.
- T. Mandai, Y. Akita, S. Yagi, M. Egashira, H. Munakata, K. Kanamura, J. Mater. Chem. A, 2017, 5, 3152–3156.
- F. Tuerxun, K. Yamamoto, M. Hattori, T. Mandai, K. Nakanishi, A. Choudhary, Y. Tateyama, K. Sodeyama, A. Nakao, T. Uchiyama, M. Matsui, K. Tsuruta, Y. Tamenori, K. Kanamura, Y. Uchimoto, ACS Appl. Mater. Interfaces, 2020, 12, 25775–25785.
- T. Watkins, A. Kumar, D. A. Buttry, J. Am. Chem. Soc., 2016, 138, 641–650.
- M. Oishi, T. Ichitsubo, S. Okamoto, S. Toyoda, E. Matsubara, T. Nohira, R. Hagiwara, J. Electrochem. Soc., 2014, 161, A943.
- S. Okamoto, T. Ichitsubo, T. Kawaguchi, Y. Kumagai, F. Oba, S. Yagi, K. Shimokawa, N. Goto, T. Doi and E. Matsubara, Adv. Sci., 2015, 2, 1500072.
- K. Sone, Y. Hayashi, T. Mandai, S. Yagi, Y. Oaki, H. Imai, J. Mater. Chem. A, 2021, 9, 6851–6860
- H. Kobayashi, K. Samukawa, M. Nakayama, T. Mandai, I. Honma, ACS Appl. Nano Mater., 2021, 4, 8328–8333.
- T. Mandai, ACS Appl. Mater. Interfaces, 2020, 12, 39135–39144.
- T. Mandai, Y. Youn, Y. Tateyama, Mater. Adv., 2021, 2, 6283–6296.