Topics
2022.03.31 circular No.18
興味の糸を繋いで紡いで独自の領域へ~しかし川の流れのように~
横浜国立大学先端科学高等研究院
渡邉 正義
大学で研究する者の幸せな点は、興味の糸を繋いで紡げることである。私自身、「興味の糸を繋いで紡いで独自の領域を作って来た」という気持ちはある。しかし同時に、時代の大きな流れがあり、その大きな流れが研究の方向を「川の流れのように」決めて来た気もする。また、大学での研究の大きな楽しみは、学生や博士研究員といった若く情熱のある方たちと、ある時は情熱を共有し、ある時はぶつかり、ある時は教え、またある時は教わるチャンスがある事である。「研究は人」と考え、学生にやる気を出させるのが一番大事であり一緒に研究を楽しむ事を心掛けてきた。一方「研究は自由、しかし能力以上の研究はできない」と言って学生を激励し、自分もそれを拠り所に過ごしてきた。ここでは長いようで短い、学生時代からの40年余りを振り返ってみたい1,2。
研究の発端
早稲田大学の学部時代は、クラブ活動の登山に明け暮れた。将来の研究に関しては、何となくエネルギーや情報に係る材料の関係が面白そうだと思っていた。卒業研究で、篠原 功先生(高分子化学)の研究室に配属され、高分子電荷移動錯体という導電性高分子の研究を始めた。同研究室で大学院に進学、学部時代の反動もあって真剣に研究に取り組みたいと思った(1978年頃)。その頃研究室には、岡野光夫氏(東京女子医大名誉教授)や石原一彦氏(東京大学名誉教授)などもいて活気に溢れていた。
当時は、2000年にノーベル化学賞を受賞された白川英樹先生が、ポリアセチレンのドーピングによる金属的な導電性を、 MacDiarmid教授、Heeger教授とともに発表した時期で、世界中がこのニュースでフィーバーしていた3。早稲田でも、白川先生がガラスアンプルに入ったポリアセチレンの実物を回覧した講演を拝聴した。私もπ-共役高分子の研究を非常に魅力的に感じ、少し方向転換をしたいと思い指導教授とも相談したが、アドバイスは予想とは全く異なるものであった。提案されたのは「これからはエネルギーが大事になるからイオン伝導性高分子の研究をやってみないか?」だった。事実その頃、ポリエチレンオキシド(PEO)とアルカリ金属塩の錯体が比較的高いイオン伝導性を発現するという報告が出され、研究の萌芽はあった4。
私はその提案を受け入れ、イオン伝導体の研究を始めたが、早大にそのような研究の蓄積はまったく無かった。そこでイオン伝導性はどうやって測るのかという所から始めた。無機の固体電解質は長い研究の歴史があるのでその文献を参考にすると、複素インピーダンス法という方法で測るということは分かった。しかし、複素インピーダンス法を理解し、伝導性を見積もることができるまでに1年位格闘した。当時は今のような電気化学インピーダンスを測る装置などは高くて手が出ず、またNyquist
plotを作成するようなソフトもなかった。LCRメータを購入して何とか凌いだ。博士課程途中(2年終了時)で、上智大学の緒方直哉先生の研究室で助手の職を得、学位論文も早稲田に提出した。
当時、緒方教授は、現在のJSTのERATO「緒方ファインポリマープロジェクト」を始めていた。このプロジェクトは新しい機能性高分子の創製に関する内容で、その中に電子伝導性高分子の研究グループがあったため、緒方先生から「研究室ではイオン伝導性高分子の研究をやってくれ」と指示され本格的な研究が始まった。しかし、当時は装置もなく電気電子工学科の研究室と装置を借りての共同研究だった。イオン伝導性高分子の研究で興味を持ったのは、高分子を従来の水や有機溶媒に代わるイオン伝導性媒体として用いたとき、どのような違いが生じ、さらにどのような事が出来るかという事であった(図1)。特に、高分子構造とイオン伝導特性、イオンダイナミックスと高分子ダイナミックスの相関、キャリヤ移動度とキャリヤ数の分離、さらにイオン輸率の問題に興味を持った5。当時研究を支えてくれた学生の一人が、陸川政弘氏(上智大学教授)であった。サイズの小さなイオンの輸送が何故高分子のセグメント運動の緩和時間の温度依存性を示すWLF式(VTF式)に従うのか、どうやったらイオン移動度やイオン輸率を測定できるかなどの課題に取り組んだ。今でもよく覚えているのが、高分子中のイオン移動度の測定を電圧極性反転法という一種のtime-of-flight法を活用して行う研究であった6。この方法は、絶縁性高分子の絶縁破壊の原因となるイオン性不純物の移動度を測定する方法として、電気工学の分野で発表があった方法である。高分子を一度直流分極してイオン性キャリヤを電極分極させ、その後反転させるという方法である。今考えると不充分な方法であるが、time-of-flightで電流ピークが観測されたときには、感激を覚えた。しかし、国内でこのようなテーマに取り組んでいる研究者は少なく、当時多くの発表をしていた高分子学会や電気化学会では閑古鳥が鳴いていた。しかし逆に多くの異分野の方と交流することができた。当時名大の高橋武彦先生は、無機固体電解質がご専門だったが、学会などでお会いすると「最近の有機系の進歩があったら論文を送って下さい」などとお声掛け頂き、励みになった。
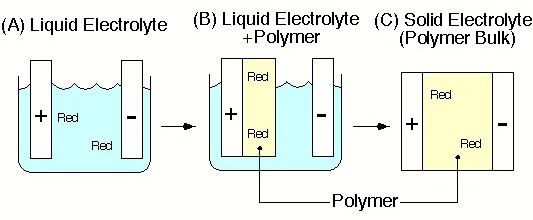
図1 これまでイオン伝導体として用いられてきた電解液の溶媒の代わりに高分子を用いたら、どうなり、何が出来るかを明らかにすることを研究目標に設定.
研究が進むと、イオン伝導のWLF型の温度依存性はイオンと高分子の強い相互作用のために、構造の緩和時間と伝導の緩和時間がカップリングしているためであると理解できた 7。しかし多くの物質群の中には構造緩和時間と伝導緩和時間がデカップリングしている系も存在し、さらにTg以上の温度域での輸送特性の温度依存性は物質のfragilityによっても変化することも分かってきた。ユニバーサルなWLF式の意味するところは、多くの非晶性高分子のfragilityは類似しているという事と理解できた。また、高分子を媒体にした固体電気化学も可能と考え、イオン伝導性高分子中にフェロセンのような分子を溶解させ、このレドックス応答をサイクリックボルタンメトリー(CV)で測定する研究を試みていた(図2)5。しかし、電解質溶液と比較すると抵抗が大きいため、なかなか綺麗なCVが得られないでいた。
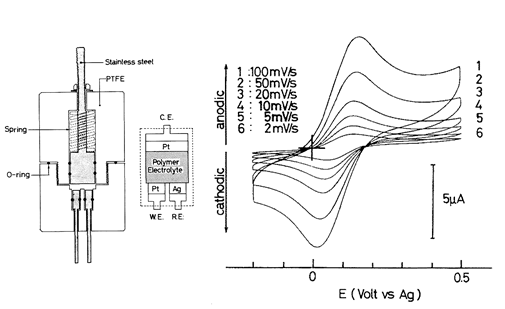
図2 イオン伝導性高分子中に溶解したフェロセンの電気化学応答を調べるために試作したセルの構造とCV(at80℃).5電解液と比べると抵抗が大きいので室温ではiRドロップが大きくて綺麗なCV測定できずに昇温して測定したが、それでも掃引速度が増すとiRドロップの影響が出ている.
Chapel Hillへの留学
たまたま関連文献を調べていると、米国North Carolina大学のRoyce
Murray教授が、PEO中に溶解したレドックス物質の電気化学応答を、マイクロ電極法を用いて調べるという研究を始めていることが分かった8。このときは、世界で同時に全く同じことを考えている研究者がいることに驚いた。今は当たり前に思うが、その当時はまだ30代初めで経験が浅かった。またMurray教授は、1970~1980年代、電気化学の世界では盛んに研究された修飾電極の権威であることも知っていた。そこでいっそのこと飛び込んで、電気化学を本格的に学ぼうと思った。早稲田も、当時在籍していた上智の研究室も、高分子化学の研究室であったため、電気化学の知識や技術は独学で培ってきたので自信がなかった。留学したChapel
Hillでの米国生活(1988-1990年)は、楽しかった(図3)。私が行った研究テーマは、iRドロップを無視できるマイクロ電極法を用いて、イオン伝導性高分子を用いた電気化学測定を実現する方法論を確立すること、この測定から高分子中でのレドックス分子の拡散係数9や均一(不均一)電子移動速度10といった特性を明らかにすることであり、私の興味と完全に一致していた。レドックス分子を高分子にすることで高分子中の高分子の拡散の問題11などにもチャレンジすることができた。
研究開始してまず行ったのが、pAレベルまで測定可能なポテンシオスタットを自作することであった。設計回路図はあったが、オペアンプを始めとする部品は全て電話で注文して揃えて、半田付けして作製した。半年近く掛かったがこれが動いたときは嬉しかった。また、pAレベルまでの測定という事で、ノイズを避けるためのFaraday
cage、XYレコーダ、そして自作のマイクロポテンシオスタットが私の研究の3種の神器となった。その後、研究は順調に進み、学生時代に取り扱ったこともあるTCNQ分子をレドックス分子に選ぶことにより、高分子相中での均一電子移動反応(電子ホッピング)に関し、興味深い結果を得た9。また、私と同時期に、西原 寛氏(東京大学名誉教授)もMurray研に在籍していて、家族ぐるみのお付き合いをさせて頂き、現在も親交は続いている。
Murray教授はJACSなどに多くの論文を出し、US Academy of
Scienceの会員でもある研究者であるが、メンバーそれぞれが自作(または学内のelectric shop作)のポテンシオスタットとXYレコーダを持っているだけで、研究室には大きな装置は殆ど無く、「研究は人」ということを強く実感した。また、英語を母国語にしている国では、英語論文作成が容易にできるのではと思っていたが、研究室にいた学生達(殆ど米国人であった)は、自分が論文草稿を書いても、最終投稿段階では殆ど自分の原稿は残らないとの事で、入念な論文作成が行われていることを実感した。この留学で、自分が独学で培ってきた電気化学に関する知識や方法論に自信が付いた。さらに、高分子中のイオン輸送だけでなく、高分子での物質拡散や電子移動などにも関心が高まり、研究の視野は著しく広がった。帰国後は次から次へとアイデアが浮かんで何でもやってみたいと思って研究の幅を広げた時期だった。この頃は、大きな国際会議の帰りの飛行機の中で何故か多くの研究のアイデアが浮かんだ。会議での知的刺激と、日本語を使わないという非日常が頭を活性化させたのかもしれない。帰りの飛行機で浮かんだアイデアは必ずメモしてお土産に持って帰った。貰った学生は迷惑だったかも知れないが・・・。

図3 米国最古の州立大学であるNorth Carolina大学のシンボルであるOld Wellとバスケットボールの専用会場であるSmith Center(人口5万人のChapel Hillに2万人以上収容の会場).
絶大な人気を誇った出身者のMichael Jordan
米国から帰国し、その後、現在の横浜国大に職を得(1992年)、まず取り組んだのが高分子中(ガラス形成液体中)でのイオン伝導の支配因子の整理であった。その結果、構造緩和時間と伝導緩和時間がカップリングしている系では、アリゾナ州立大学のAusten
Angell教授が提唱していたfragilityが大きくなると、Tg以上の温度域でのイオン伝導性も増大する(図4)12。また構造緩和時間と伝導緩和時間をデカップリングさせることが出来れば、Tg近くになっても高い伝導性を維持できるという事であった。ガラス状態でも高い伝導性を示す超イオン伝導性ガラスなどでは、Tg以上の温度域でも構造緩和時間と伝導緩和時間が大きくデカップリングしていることも知った。
横浜国大に異動して、「研究をやるぞ!」という気合は入っていたが、実験室は整備されていない、スタッフはいない、お金は無いところから始まった。しかしこちらの情熱が伝わったのだろうか、上智の修士から横浜国大の博士に入学してくれた長坂秀昭博士(元キヤノン)、博士課程まで進学してくれた異動2年目の学部生、西本 淳博士(Northvolt)、齋藤貴宏博士(三菱ケミカル)、中山大輔博士(富士フィルムビジネスイノベーション)らと一緒に、何とか研究室を立ち上げた。彼らの存在は、研究室立ち上げ期に非常に大きかった。
横浜国大では、主鎖セグメント運動より速い側鎖の運動とイオン輸送をカップルできないかという課題にまず挑戦した。色々な側鎖分岐ポリエーテルを合成し、現在でもポリエーテル系では最も高いイオン伝導体(> 10-4 Scm-1@ RT)を実現した13。Tgが同一でも、分岐側鎖を導入すると伝導性が増大することから、速い側鎖の運動がイオン輸送に寄与した結果と結論した。この研究に大きく貢献してくれたのが、西本 淳博士や社会人博士であった河野通之博士(元第一工業製薬)である。今考えると、この結果は、高分子のfragilityを変化(増大)させた結果とも考えられる(図4)。また、ポリエーテル中でのリチウムイオン輸率を増大させるため、ルイス酸性基を導入してアニオンの移動度を低下させたり、高分子リチウム塩とポリエーテルのポリマーアロイ電解質の検討を、田畑誠一郎博士(ソニー)を中心に行った。しかし、ポリエーテル系の高分子ではキャリヤイオン数を増大させるとTg が上昇してキャリヤ移動度が低下する問題から脱却することができないため、イオン伝導性は溶液レベルに到達しないと結論していた。
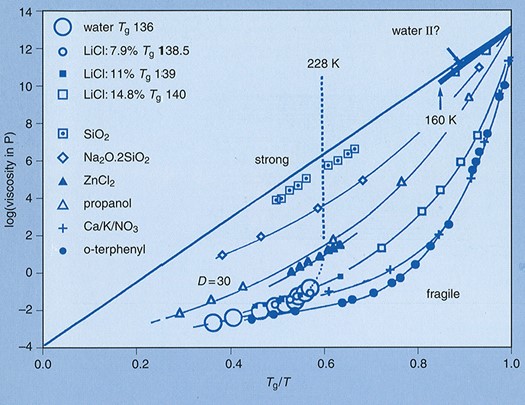
図4 ガラス形成液体の粘度のTg で規格化したArrhenius plot (Angell plot).12Tgで規格化することにより全ての物質の粘度の温度依存性が1つの図で比較できる.温度依存性が直線に近い物質をStrong,撓みの大きな物質をFragileな物質という。粘度とカップリングしたイオン輸送が起こる系では、よりFragileな物質が高い伝導性となる.
一方、米国から帰国した年に参加した国際会議の帰りの北京空港で、その当時ガラスを専門としていた河村純一氏(東北大特任教授)と飛行機待ち時間に雑談した(1990年)。ガラスと高分子の比較の議論から「高分子に複合化するとTgが低下するような塩はないかな?」という話をすると「室温で液体の常温溶融塩というものがあるらしいよ」との答えだった。早速調べてみると対アニオンがクロロアルミナート(AlCl4-やAl2Cl7-)のイミダゾリウム塩やピリジニウム塩が室温で液体であると分かった。しかしこれらの原料となるAlCl3は、灰色でいかにも純度の低いものしか手に入らなかった。そこで、クロロアルミナート系常温溶融塩で実績のあったニューヨーク大学バッファロー校のRobert Osteryoung教授を訪問し、直々にAlCl3の精製法をご教授頂いた(図5)。精製したAlCl3を用い、高分子との複合化を試みた。この研究は上智で開始し、山田心一郎氏(Dexerials)が頑張ってくれた。この系は常温溶融塩の組成が高くなるほど系のTg が低くなり、キャリヤ濃度と移動度増加が同時に達成できるため、室温で10-3Scm-1 のイオン伝導性に到達したと1993年に報告した14。同時期にAngell 教授のグループからTgの低いLi塩混合物とポリエーテルの複合系が、高いイオン伝導性を示すpolymer-in-salt電解質として報告され15、研究は活性化した。
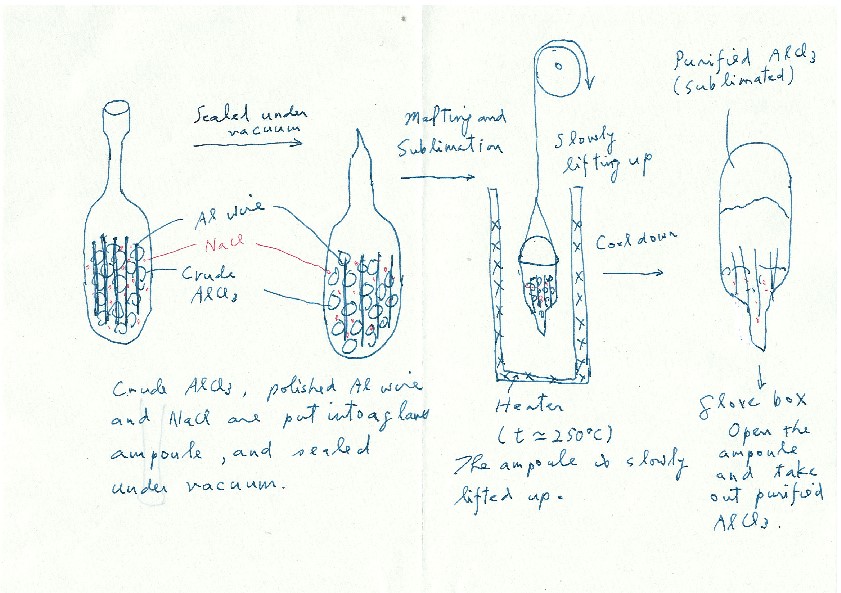
図5 Osteryoung研究室を訪問したときにご教授頂いたAlCl3精製法を記したメモ.
しかし、AlCl4-などをアニオンとするクロロアルミナート系溶融塩の泣き所は、水分に非常に敏感で分解し易いことであった。丁度その頃、水分にも酸素にも安定な常温溶融塩が見出され、イオン液体とも呼ばれ研究が活性化し始めていた。我々も自然に興味を持ち研究を進めた。クロロアルミナート系溶融塩との違いは、対アニオンがより安定な、BF4-や[N(SO2CF3)2]-などを用いる事であった。まずはイオン液体の基礎物性をきちんと見極めたいと思った。そんな時、パルス磁場勾配NMR(PFG-NMR)法で有機電解液中のイオンの自己拡散係数を測定していた元産総研の早水紀久子博士の研究に出会った。これを是非イオン液体に適用したいと思った。電子伝導性物質でキャリヤ移動度を測る手法は種々あるが、イオン伝導性物質でキャリヤ移動度を正確に測る手法はなかった。自己拡散係数が計測できれば、イオン伝導性をキャリヤ移動度とキャリヤ数に分離できる。大学にPFG-NMRの装置は無かったので、つくばの産総研の装置をお借りし、学生に泊り掛けで拡散係数を測ってもらった。このとき大活躍してくれたのが、野田明宏博士(本田技研)や徳田浩之博士(三菱ケミカル)であった。その後、大型の科研費を獲得することができ、大学にPFG-NMRの装置を導入することができた。さらに早水先生が学生を丁寧に指導して下さり、イオン液体の研究を本格的に始める体制が整った。2000年を過ぎた頃の事である 16。
研究の幅を広げ研究室スタッフと協働
横浜国大に着任してからの研究テーマには、上記イオン伝導体以外に、高分子バルク中での均一電子移動、ハイドロゲルの膨潤収縮のダイナミックスや分子認識の可能性を取り上げた。
電子移動のテーマは、イオン伝導性高分子中に共有結合で固定化したレドックスサイト(フェロセン、フェノチアジン等)間をどのように電子移動反応が起こるかという興味に基づいていた17。すなわち、これが可能になれば低分子溶媒を用いないall-in-one
electrochemistryが実現できると考えていた。これは長坂博士が担当してくれた。その結果、巨視的に動けないレドックスサイト間でも電子移動反応が起こり、電気化学の理論家であったJean-Michel
Savéant教授が提案した、束縛拡散モデルで説明できることが分かった。レドックスサイトは高分子鎖に固定されていても局所的に分子運動(束縛拡散)可能であれば、その結果電子移動反応が起こるという内容であった17。従って、電子移動速度はレドックスサイトの濃度、高分子のダイナミックス(Tgとの温度差)に大きく影響を受けることが分かった。この研究は、齋藤博士に引き継がれ、電子移動反応を用いて、酵素反応のメディエータとして用いる研究に発展した。
一方、高分子ハイドロゲルの研究は、中山博士が担当してくれた。マイクロディスク電極上にハイドロゲルを化学固定化し、これが膨潤収縮する際のレドックス物質のゲル相内の拡散係数や濃度を求めて、ゲルの膨潤収縮のダイナミックスを研究した。さらに、ハイドロゲルに分子認識能を持たせることが可能かなどの課題を検討した18。
1998年、教授に昇任させて頂き、今林慎一郎氏(芝浦工業大学教授)、竹岡敬和氏(名古屋大学准教授)と一緒に研究室を運営することになった。今林先生は、横浜国大の仁木克己先生の研究室ご出身で、生物電気化学関係に関心があった。そこで米国での研究が発端になった、レドックス活性基(フェノチアジン)を片末端に有するPEOを酵素や電極表面に修飾した生物電子移動反応の研究を共同で進めた。興味深かったのは、レドックス活性基を介した酵素反応のメディエーションが、PEOスペーサ長や修飾位置によって、メディエータ共存系より迅速になることであった(図6)19 。この研究では、齋藤博士や修士時代の上木岳士博士(NIMS)が活躍してくれた。後述するが上木博士は修士修了後、企業に就職し、もう一度博士課程に入り直してくれて、イオン液体の研究で活躍してくれた。
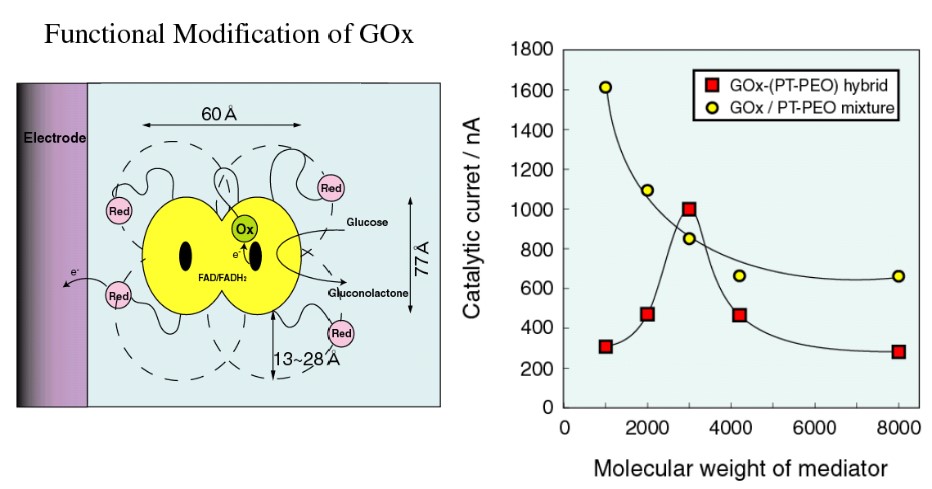
図6 グルコース酸化酵素のリシン残基にPEO鎖を介したフェノチアジン(PT-PEO)を修飾したハイブリッド(左図)の触媒酸化電流のPT-PEO分子量依存性(右図).触媒反応に最適PEOスペーサ長があり、そのときの活性はPT-PEO混合系より高くなる.19
竹岡先生は、上智の緒方先生の所で学位を取得した。博士課程の頃から、私との共同研究を始め、レドックス活性界面活性剤の分子集合状態とレドックス応答、さらにそれを薬物徐放に展開する研究を行っていた。学位取得後、高分子ゲルの研究で著名であった、MITの田中豊一先生の所での留学から帰って来て、助手になって頂いた。初めは、研究室のゲル研究を手伝って頂いていたが、シリカ微粒子からなるコロイド結晶の隙間で合成し、シリカを除いたインバースオパール構造を有する多孔ゲルが構造色を示すことを発見し、ゲルの膨潤収縮によって構造色が変化することを報告した。この研究は、中山博士の研究と合流し、分子認識構造色センサーの研究へと発展した(図7)20。
この時期(1992年~2002年頃)は、横浜国大に赴任してからの10年間で、研究室の整備とともに、米国留学で広げた関心に従って研究の幅を広げたとも言えるし、また自分の研究の方向性を模索して、もがいていた時期とも言える。
図7 フェニルボロン酸を導入したグルコースに応答して膨潤度変化するゲルに、インバースオパール構造を持たせることによる、グルコース濃度に応答した構造色変化. 20
イオン液体の本格的な研究前述したイオン液体の基礎物性理解に関しては、イオン液体がどうして溶媒無しに電離液体として振舞うのかという点を解明したかった。そこで、PFG-NMRを利用してその輸送現象を調べた。イオン伝導率測定から求まるモル伝導率とPFG-NMRから測定されるカチオン、アニオンの自己拡散係数とNernst-Einstein式から求まるモル伝導率の比をイオン性 (ionicity)と定義して、多くの汎用イオン液体について系統的に調査した21,22 。その結果、ionicityは、カチオン、アニオン間のクーロン相互作用と、イオン間のvan der Waals力の微妙なバランスに影響されることを明らかにした(図8)。特に、van der Waals力が関与するという特徴は、従来研究されていた高温溶融塩には無い事実であったので、注目された。この研究で中心的役割を果たしてくれたのが、徳田博士である。またこれらの研究は、都築誠二氏(産総研つくば)、篠田 渉氏(岡山大学教授)らによる計算結果とも対応させた。この関連の原著論文4本が1000回以上引用されている。
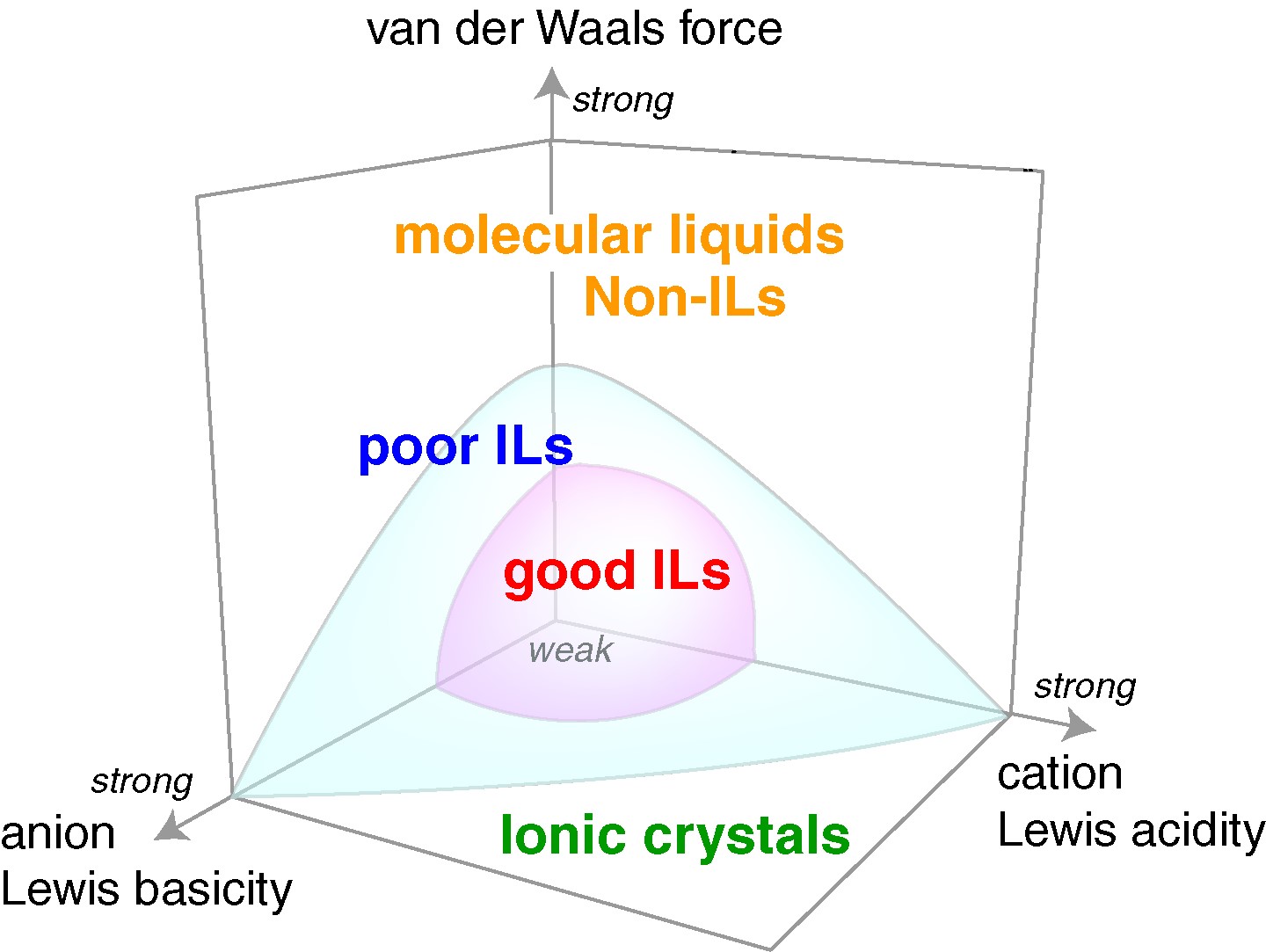
図8 イオン液体のionicityを決めるアニオンのLewis塩基性、カチオンのLewis酸性、イオン間のvan der Waals力.22 Good ILs: ionicityが1に近いILs;poor ILs: ionicityが1より低いILs.
クロロアルミナート系イオン液体と高分子からなるイオン伝導体の研究は、水や酸素にも安定なイオン液体を用いた研究に発展した。特に、ビニル系モノマーをイオン液体に溶かし、そのまま重合してゲル化でき、重合後も相溶している系をイオンゲルと名付けて研究を進めた(図9)18。前述した野田博士や、M.A.B.H. Susan博士(ダッカ大学教授)がこの研究を進めてくれた。さらに、イオン伝導性増大のためのもう一つの方法論と考えていた、構造緩和時間と伝導緩和時間のデカップリングはこの系で証明した。従来のポリエーテル系電解質と比較したときの顕著な違いは、イオン液体の高分子に対する可塑化効果とセグメント運動からデカップリングしたイオン輸送であった(図10)24 。この研究のためには広温度範囲(-70~130℃)でのイオン伝導性の精密測定が必要で、関 志朗博士(工学院大学准教授)が徹夜で頑張ってくれた。その結果、電解質溶液に匹敵するイオン輸送性が実現し、同時に不揮発性と熱安定性はイオン液体の特性から確保された。さらに高分子の化学構造は、イオン液体のionicityに影響を与えることも見出した。
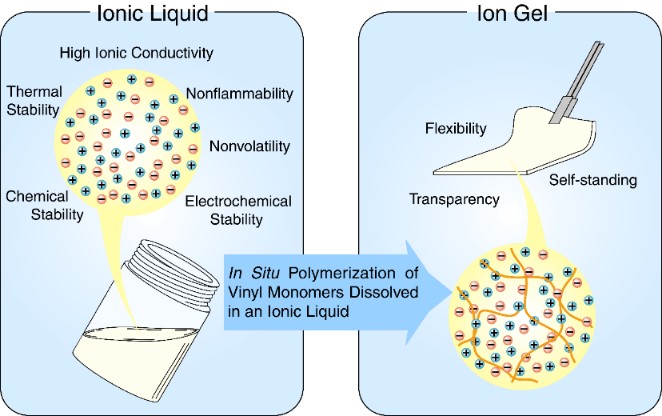
図9 イオン液体中のモノマーのin situ重合によって得られるイオンゲル.18
高分子以外でイオン液体を固体化する方法として、ナノ粒子の添加がある。その当時、イオン液体中でナノ粒子(金属、半導体、酸化物等)を合成したり、安定分散させたりという研究が散見された。しかし、イオン液体はイオン強度が高いため、常識的にはコロイド分散のための良い分散媒とは考え難かった。そこで上野和英博士(横浜国大准教授)の博士課程のテーマに、イオン液体中にナノ粒子を分散させた系の検討を提案し、彼が系統的に研究を進めてくれた25。モデルとしてシリカナノ粒子を選択して検討を加えた。その結果、やはり一般的にはイオン液体中にナノ粒子は分散し難く、凝集してネットワークを形成してゲル化する。この現象を利用すると、5wt%程度のシリカ粒子の添加でゲル化が始まり、15wt%程度添加するとイオン液体が粉末になる事を見出した。しかし、このような大きな巨視的物性の変化に対し、系のイオン伝導性に殆ど変化がなく、イオン輸送と力学特性が大きくデカップリングしていることが分かった。この現象を利用すると、イオン液体を用いた固体電解質の創製が可能となり、ある企業による大型電池への展開も行われた。このような凝集現象が一般的であるが、ナノ粒子表面に対して特異的相互作用のあるカチオンあるいはアニオンがあると表面吸着し、さらにクーロン相互作用によって反対イオンが吸着し層状構造を形成して凝集し難くなる場合があることも分かった。溶媒和力と言われている分散機構である。さらにイオン液体に相溶性の高い高分子をナノ粒子上にグラフト化すると安定分散し、一定濃度以上でコロイドガラス化することも上野博士によって見いだされた。このコロイドガラスは、シリカ粒子径を上手く選択すると可視光を選択反射する構造色を呈することも見出された。
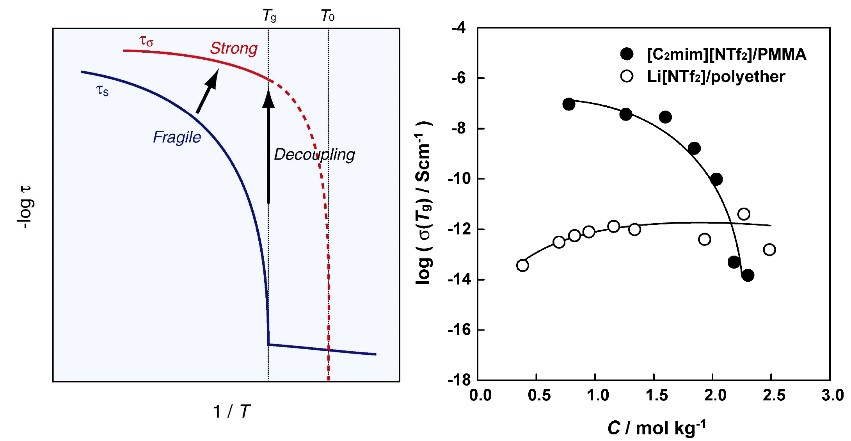
図10 構造緩和時間(ts)と伝導緩和時間(tσ) のデカップリングの概念(左図).ポリエーテル中のイオン伝導がカップリング輸送であるのに対し、イオンゲル中でのデカップリング現象. 24アメリカ化学会の許可を得て転載(右図).
イオン液体の基礎物性および高分子やナノ粒子を複合化したイオンゲルのデカップリングしたイオン輸送を明らかにすることができたので、その後の研究は機能性イオン液体の創製に向かった26。その一つが、プロトン性イオン液体の物性評価と無加湿中温型燃料電池への展開である27。プロトン性イオン液体(PIL)とは活性プロトンを有するイオン液体で、最も広く用いられている方法は、ブレンステッド酸と塩基の間のプロトン移動反応による塩形成である。このプロトン移動反応は平衡反応であり、1か0かという話でない事が面白かった。普段あまり気に留めないが、アンモニア水溶液は塩基性になるが、水酸化アンモニウムは単離できないことが良い例である。基本的には酸とプロトン化された塩基の間のΔpKaがプロトン移動の駆動力として重要な役割を果たすことが、梅林泰宏氏(新潟大学教授)らによっても指摘されている。実際に典型的な非プロトン性イオン液体(APIL)並みの耐熱性を備えるためにはΔpKa>15、さらに酸・塩基の化学構造にも影響を受けるという結果を得ている。この研究は、M.S. Miran博士(ダッカ大学教授)らによって進められた。さらにPILのionicityは、典型的なAPILのそれより低く、これまで検討した系では0.5程度が最大になる。構造異性体のPILとAPILを用いてカチオン・アニオン間の相互作用エネルギーを計算すると、10 kcal/mol程度PILの方が大きくなり、これはカチオンのNHとアニオンの間の水素結合が存在するためであることが都築博士らによって明らかにされている。このようなPILが、プロトン伝導体として機能するのではと着想し、野田博士らによって研究開始された。当初はH[NTf2]とイミダゾール(Im)からなる二元系の相図や輸送特性を検討した。イオン伝導性の極大が、Im過剰な組成で認められ、これはGrotthuss型のプロトン輸送によることが分かった。また、これらの液体を電解質に用いて、液体型の水素・酸素燃料電池が無加湿下で作動することを見出した。しかし、中温型燃料電池(100~150℃程度)に展開するためには、耐熱性の観点から酸・塩基1:1の組成のPILでなければ中性分子が蒸発してしまうことも分かった。そこで100種以上のPILを合成して、その特性、特に水素酸化、酸素還元といった燃料電池反応の特性を調べてくれたのが中本博文博士(トヨタ自動車)であった。この探索研究のなかには、濱口宏夫氏(東京大学名誉教授)との共同研究もある28。その結果、[(C2H5)2CH3NH][CF3SO3]([dema][TfO])が優れた特性を示すことを見出した。現在、この物質が燃料電池適用を目指したPILの世界標準になっている。この結果を得て、[dema][TfO]を何とか膜型燃料電池に展開できないかと模索した。イオン液体を担持できて、かつ膜型燃料電池に適用できるような力学強度を備える膜は中々得られない。試行錯誤の結果、スルホン化ポリイミドが相溶性、力学強度、電気化学特性などの特性を備える事を見出した。スルホン化ポリイミドは他のアイオノマーと同様に、イオン部分と非イオン部分が相分離した構造を形成し、イオン液体はイオン部分に選択的に取り込まれ、かつイオン部分と非イオン部分が共連続構造を形成することが、輸送特性と力学特性を両立させる鍵と分かった。この複合膜を用いることにより、無加湿中温型燃料電池が構築できることを示した(図11)29。この研究は、博士研究員の安田友洋氏(三菱マテリアル)、李 承烈博士(釜山大学校)が頑張ってくれた。スルホン化ポリイミドの薄膜成型性は、最近のCO2選択透過膜の研究にも発展している。
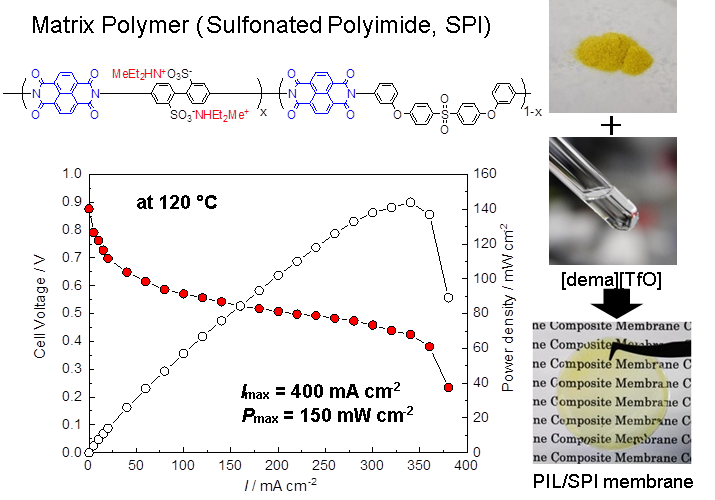
図11 プロトン性イオン液体([dema][TfO])とスルホン化ポリイミドからなる複合膜と、これを用いた無加湿中温形燃料電池の分極曲線.2
またPILを新しい材料創製の原料にする研究も進んだ。PILの熱重量分析をすると、中には高温での残渣の量が著しく多い系が存在することはMiran博士の研究で分かっていた。この点に着目し、新しい炭素材料の研究に展開したのが博士研究員の張
世国氏(湖南大学教授)、Mahfuzul Hoque博士(U. British
Columbia)であった。PIL、特に硫酸水素塩、を無酸素下で炭化すると種々の高電子伝導性N-doped
Carbonが得られ、酸素還元触媒能、電気二重層キャパシタ特性、CO2吸着能などの優れた材料創製に繋がった30。
二番目の機能性イオン液体として、電子輸送性イオン液体の研究を行った。この研究は、柳田祥三氏(大阪大学名誉教授)と(株)フジクラと共同で色素増感太陽電池(DSSC)構築を目的に行われた。DSSCはGrätzelセルとも呼ばれる湿式太陽電池で、色素増感されたTiO2フォトアノードで酸化反応が、対極で還元反応が起こる。光励起されてできた色素のホールによって酸化反応が起こる必要があるので、電子輸送を担うレドックス対としては電位の観点から、I-/I3-が良く用いられる。これらは電解液中を迅速に輸送されることが重要であると同時に、電解液は揮発性が低く熱安定性に優れることが要求された。そこでイオン液体を研究していた私にお呼びが掛かった訳である。マイクロ電極を用いてI-/I3-のイオン液体中での輸送特性を測定したところ、このレドックス対の濃度増大に伴い、拡散係数が増大することを見出した31。すなわち、レドックス対の濃度を上げると電子輸送性が高くなるということである。一方、分子性溶媒中ではこのような現象は現れなかった。イオン液体中でのこの現象は、I-とI3-の間でI2の交換反応による構造拡散が起こっているためと考えられた。I-とI3-の間の二次反応であるこの交換反応は、アニオン同士の静電反発のため、一般的には起こり難い。しかし、イオン強度の高いイオン液体中では静電遮蔽が起こるために、促進されると結論した。一種のkinetic
salt
effectと考えられる。この研究で中心的に活躍してくれたのは、川野竜司博士(東京農工大学教授)、片伯部 貫博士(エプソン)である。イオン液体電解質は粘度が高いが、DSSCに適用すると、汎用の有機電解液に匹敵する特性が発現した。このイオン液体中でのkinetic
salt effectに関しては、髙橋憲司氏(金沢大学教授)とも放射化学を使った研究で共同させて頂いた32。川野博士は、ヒューマンメディエータ能力が高かった。柳田先生の研究室とは、スメクチック構造を有するイオン液体液晶を電解質に用いるとI-/I3-がスメクチック層間に濃縮されるため、構造拡散がさらに効率的に進むことを共同研究で進めてくれた33。また、イオン液体電解質にシリカナノ粒子を添加すると、ゲル化するとともにI-/I3-の輸送速度も速くなることを、片伯部博士が見出した。I-/I3-がシリカ表面にカチオンを介して特異吸着し、構造拡散のハイウエーを形成した結果と考えた。さらに、高分子イオン液体を用いた電解質を用いてフレキシブル電池が構築できることをGrätzel先生と共同で発表することができた(図12)34。
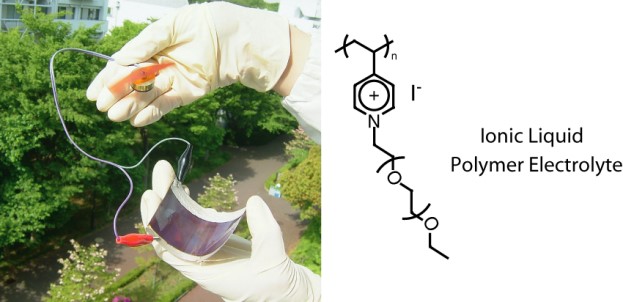
図12 高分子イオン液体を用いたフレキシブル色素増感太陽電池の構築.34
三番目の機能性イオン液体として、リチウムイオン伝導性イオン液体の研究を進めた。イオン液体の研究が活発化した当初から、イオン液体をリチウム電池電解質に適用することは世界中の多くの研究者の関心の的であった。現在、最高のエネルギー密度を有する二次電池はリチウムイオン電池 (LIB)であるが、その使用範囲や規模の増大に伴って、有機電解液を用いることによる安全性への懸念があるためである。また、LIBよりさらにエネルギー密度を増大させるために金属リチウム負極の利用が期待されているが、安全性の問題はLIBより深刻になる。我々も含め多くの研究者は、イオン液体中にリチウム塩を溶解した電解質の研究を始めた(図13(a))。同時に、リチウムイオンをカチオンとするイオン液体が出来ないかという課題に挑戦した。その結果、ボレートアニオンに電子求引基とPEOを導入する事によって、リチウムイオン液体を実現することができた(図13(b))。この研究は、菖蒲川 仁博士(旭化成)が頑張ってくれた。しかしその輸送特性は、通常の有機電解液と比較すると遥かに劣っていた。
その結果生まれたのが、トリグライム(G3)やテトラグライム(G4)などのグライム類とLi[NTf2]などの1:1混合物である溶媒和イオン液体である(図13(c))35,36。この混合物が、イオン液体類似の性質を表すためには、[Li(G)]+が安定で寿命が長く、独立したイオンとして振る舞う必要がある(図14)。この混合物中では、Lewis酸であるLi+に対する、Lewis塩基であるグライム(G)とアニオンの配位競合がある。溶媒和イオン液体が形成されるためには、グライムの配位による安定化が、Li+とアニオン間相互作用より優位になる必要がある。従って、アニオンの構造、グライムの構造によって溶媒和イオン液体が形成されるか否かは大きな影響を受ける。この研究には多くの博士課程学生が貢献してくれ、田村 崇博士(ソニー)、吉田和生博士(BASFジャパン)、張 策博士(CAST)などが中心となり進めた。また研究室内外の共同研究者の貢献も大きかった。研究室のスタッフになって頂いた上野博士にはPFG-NMRを用いた、梅林先生にはラマン散乱を用いた、都築先生には計算科学を用いた、溶媒和イオン液体形成判定に関して貢献頂いた。溶媒和イオン液体は海外の研究者も注目してくれて、J.
N. C. Lopes氏(Tech. Univ. of Lisbon 教授)、Rob Atkin氏(Univ. Western Australia
教授)などとの共同研究も進んだ。我々が、グライム錯体と呼んでいたこの液体に、溶媒和イオン液体の命名をしてくれたのはAngell教授であり37、世界的に広 く市民権を得た感があり、感謝している。
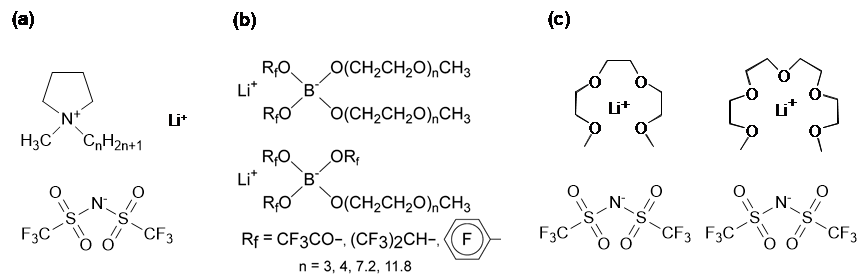
図13 リチウムイオン伝導性イオン液体創製の方法論.
グライム系溶媒和イオン液体には多くの興味深い特徴があることを見出した。
1) イオン液体類似の性質
2) 配位子であるグライム類の酸化安定性の増大
3) 他の電解液には見られないイオン性化合物の低溶解性(弱配位性)
4) 自由溶媒の活量低下さらには溶媒和イオンの不安定化による特異な電気化学反応
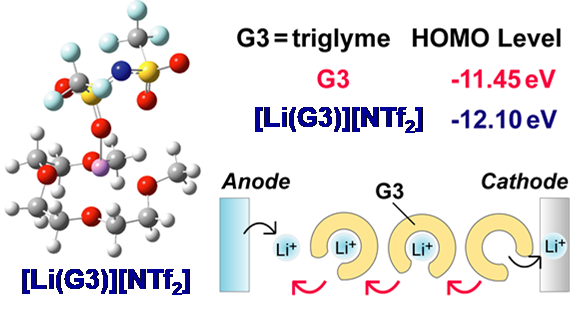
図14グライム系溶媒和イオン液体の構造、電子状態、および輸送機構の概念図.36
1)に関しては前述のように、この液体が基本的に、[Li(G)]+カチオンとアニオンから成ることに由来する。2)はグライムの酸素原子の孤立電子対がLi+に配位し、その強い電場効果を受けるために起こる。グライムの酸化はこの孤立電子対から電子が引き抜かれることによって開始するが、Li+の強い電場効果によって引き抜かれ難くなる(HOMOレベルの低下)ため酸化安定性が増大する(図14)。この事実を端的に示したのが、グライム錯体を電解質に用いた、LiCoO2の充放電の可逆性の提示であった。LiCoO2はLIBでも用いられているカソード材料で、通常4.2 V vs. Li/Li+まで充電される。グライム等のエーテル類は還元には強いが、酸化には弱いため、これまでLiCoO2 等の4 V級カソードを用いる電池の電解液には用いられてこなかった。グライム錯体を用いて、LiCoO2の可逆的充放電を示した訳である。この可逆な充放電が起こるためにはLiCoO2放電反応(Li+挿入反応)に伴って生成した自由なグライムが配位子交換をして伝導に寄与することも見出した(図14)。3)もイオン液体類に見られる興味深い性質である。通常電解液に用いる溶媒は、電解質塩を充分に溶解・解離させるため、高誘電率溶媒が用いられる。従って、他のイオン性物質に対する溶解性も高い。しかし、イオン液体を構成する多くのイオンは、カチオンのLewis酸性もアニオンのLewis塩基性も低いため、イオン性溶質の溶解性は低くなる。溶媒和イオン液体の場合には、グライムが全てLi+に配位し、自由な溶媒が殆ど無いためイオン性溶質の溶解性は低くなるとも考えられる。この事を利用して、放電生成物の溶解が深刻な問題であるリチウム硫黄電池やリチウム空気電池といった次世代二次電池電解質に適用することを着想した。リチウム硫黄電池の研究には、片山 靖氏(慶應義塾大学教授)の研究室で学位を取った立川 直樹氏(香川高専)が博士研究員として、さらに朴 埈佑博士(KERI)、Zhe Li博士(GM)などが貢献してくれた。リチウム硫黄電池に適用する研究は、JSTのプロジェクトにも取り上げられ、現在も継続している。また、リチウム空気電池の研究には、Morgan L. Thomas氏(上智大准教授)が博士研究員として、また多々良涼一博士(東京理科大助教)、 權 會旻博士(現代自動車)が頑張ってくれた。4)の例は、グラファイトの電気化学インターカレーション反応に見られる。この反応は現在LIBの負極反応にも用いられているが、電解液中の溶媒和イオンがそのままインターカレートする(共挿入反応と言われる)とグラファイトの層間剥離が進行してしまい、負極として機能しなくなる。この共挿入反応を抑制するためには、溶媒の還元分解で生成する不働態膜が不可欠と考えられ、これを生成するためには炭酸エチレン等の溶媒が必要と考えられてきた。ところが、グライム錯体を用いると、グラファイトの可逆的な電気化学インターカレーション反応が進行した。興味深いことに、グライムのLi[NTf2]に対するモル比が1を超えると(グライム過剰)共挿入反応が起こってしまうことが分かった。我々以外のグループでも、濃厚電解液を用いるとグラファイトの可逆な電気化学インターカレーション反応が起こることが報告された。この研究は、今林先生の後継として研究室のスタッフになって頂いた獨古 薫氏(横浜国大教授)が、文 善俊博士(LGエレクトロニクス)や多々良博士らと検討を進めてくれた。グライム錯体のように濃厚電解液となり自由溶媒の活量が低下すると、溶媒和イオンが不安定になり、脱溶媒和しやすくなることが一因と考えられている。また獨古先生は、Li+以外のNa+, K+やMg2+などのイオンの溶媒和イオン液体の研究を、西川恵子氏(千葉大学名誉教授)の元で学位を取得して博士研究員となった万代俊彦氏(NIMS)や、寺田尚志博士(パナソニック)などとともに発展させた。
イオン液体と高分子を用いた刺激応答材料イオン液体と高分子の複合化に関しても興味深い研究方向が見つかった。元々イオン液体への関心は、高分子との複合化にあったので、イオン液体中への高分子の溶解性については広範に検討した38。イオン液体と相溶する高分子の研究展開に関し、前述のスルホン化ポリイミドを用いた複合膜は、無加湿燃料電池の電解質膜をきっかけに、アクチュエータやCO2分離膜の研究に発展した。また溶媒和イオン液体を用いたイオンゲルは、耐熱性、難燃性の固体電解質として、リチウム系二次電池に展開可能であることを示した。ここでは、イオン性高分子アクチュエータの研究について少し詳しく述べる。この研究は日本にオリジナリティーがあり、Nafion膜の両側に無電解メッキで白金を析出させた膜が、加湿下で電圧印加によって屈曲運動することが報告された39。構造としては、高分子の電解質膜を用いた電気二重層キャパシタと同じということになる。しかし、このようなイオン性アクチュエータはイオンが電場下で泳動することを運動の原理としているために、水中や充分に加湿した条件では可動するが、乾燥雰囲気や真空中では機能しない。そこで蒸発しないイオン液体の活用が始まった。我々は、ABAブロック共重合体を用い、Aブロックにイオン液体に不溶な高分子、Bブロックにイオン液体と相溶する高分子を選択して研究を開始した(図15)40。この系は、Bブロックに選択的にイオン液体が溶解してイオン伝導パスを形成し、Aブロックが凝集して架橋点として働く熱可塑性弾性体となる。また電極には活性炭とイオン液体を含有する高分子を用いた。さらに屈曲運動機構を調べるために、イオン液体相溶ブロックにポリエーテルを選択し、ドープする電解質としてイオン液体に加えて、Li塩を用いた。この選択は、それまでの研究を大いに反映していて、Li塩でカチオン輸率が0.5以下であるのに対して、イオン液体では0.5以上であることが分かっていた。さらにイオンの大きさもこの両者で違う。その結果、電場印加によって屈曲運動する方向の異なるアクチュエータを実現できた。アクチュエータの屈曲運動は、イオン輸率とイオンの大きさの違いによる、電極層の体積変化に非対称が現れるためであることを証明した。この研究には、今泉 暁博士(広栄化学)が大きな貢献をしてくれた。その後、電極層に用いる炭素種の影響や、poly(ionic liquid)の活用の研究に発展し、竹岡先生のあとに着任した、小久保 尚氏(横浜国大特別研究教員)が、これらの研究も含め高分子合成の係る研究に貢献した。
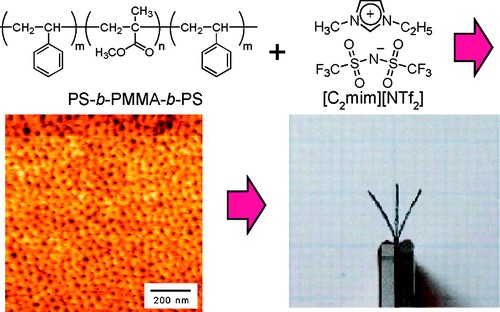
図15 イオン液体とpolystyrene-b-poly(methyl methacrylate)-b-polystyrene ABAブロック共重合体の複合膜を用いた高分子アクチュエータ.40 アメリカ化学会の許可を得て転載.
上記のような、イオン液体と高分子の相溶系のみならず、温度を上げると溶けたり(上限臨界溶液温度型、UCST型)、逆に溶けなくなったり(下限臨界溶液温度型、LCST型)という現象を見出した(図16)41。この研究の基盤を作ってくれたのが、博士課程に入り直してくれた上木博士である。特にLCST型は、構造形成性溶媒和(ΔHmix< 0, ΔSmix<0)を伴い、水以外の溶媒中の高分子で観測されることは極めて珍しいので興味を持った。イオン液体中でLCST型の相分離を示す高分子として、ポリベンジルメタクリレート(PBnMA)およびこの誘導体である側鎖にフェニル基を有するメタクリレート類、ポリエーテル等を見出した。この溶媒和による構造形成は興味深く、PBnMA類ではイオン液体のイミダゾリウム系カチオンと高分子側鎖のフェニル基との間のカチオン-π相互作用、ポリエーテルでは、芳香族カチオンの酸性プロトンとエーテル酸素の間の水素結合が、構造形成性溶媒和を形成していることが分かった。藤井健太氏(山口大学教授)、柴山充弘氏(東京大学名誉教授)らと共同で、高エネルギーX線回折と分子動力学シミュレーションによりPBnMAの溶媒和構造を検討した結果、イミダゾリウムカチオンはフェニル基を挟むように規則配列しているのに対し、[NTf2]-アニオンはフェニル基のエカトリアル位に幅広く分布していることが示された42。また、LCSTの相分離温度(Tc)は、高分子構造、イオン液体構造、さらにイオン液体ブレンドによって敏感に変化することを見出した。この理由を探る目的で、西川先生と共同で、PBnMAのイオン液体溶液の高感度DSC測定を行った。その結果、溶解に伴うΔHmix<0, ΔSmix<0を証明することができ、さらにこれらパラメータの絶対値が、典型的なLCST型相分離を起こす水溶性高分子などと比較して、1/10程度と非常に小さいことが分かった43 。その結果、ΔHmix/ΔSmixで決るTcが、高分子構造、イオン液体構造の微小な変化で大きく変化すると結論された。高分子の化学構造やイオン液体構造とTcの関係については、小玉康一氏(埼玉大学准教授)が博士研究員として系統的に研究を進めてくれた。
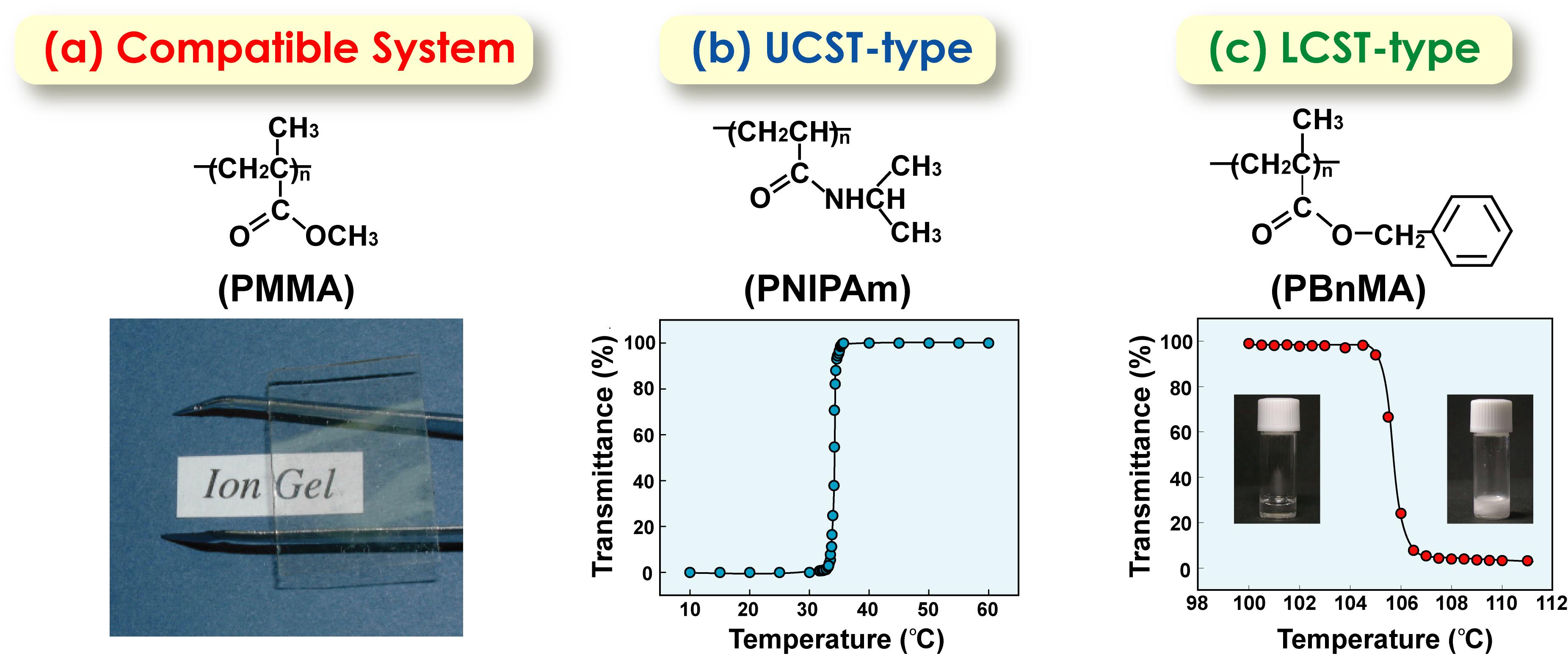
図16イオン液体([C2mim][NTf2]中に完全相溶するPMMA, UCST型相分離をするPNIPAm, LCST型相分離をするPBnMA. 41 アメリカ化学会の許可を得て転載.
高分子のUCSTおよびLCST型の相分離のTcが高分子やイオン液体の構造の僅かな変化で大きく変わる現象は、新規刺激応答材料の創製に使えると着想した。まず初めに取り組んだのが、高分子を三次元網目構造にしたゲルの膨潤・収縮応答である。PBnMAを用いたゲル(非イオン性ゲル)の体積相転移を、イオン液体中で初めて示すことができた。ゲルの膨潤・収縮は基本的に高分子鎖の共同拡散で起こるため、粘度が通常の溶媒より高いイオン液体中では変化が非常に遅い。この体積相転移を見出した上木博士は、2晩位泊り掛けで実験してくれた。
このイオン液体と高分子を用いた刺激応答材料の研究は、Timothy Lodge氏(ミネソタ大学教授)が注目してくれた。当時Lodge先生はACSのMacromoleculesのEditor-in-Chiefをしていて、我々にイオン液体と高分子のレビューを書くことを薦めてくれた41。これがきっかりでお付き合いが始まった。当時Lodge先生は、イオン液体中でブロック共重合体が作る自己集合の研究を始めていて、我々が発見したUCSTおよびLCST型の相分離と組み合わせることに興味を持ったと考えられる。丁度、博士号を取った上木博士が、ポスドク先としてLodge研を選び、共同研究を進めるに至った。上述したゲルと異なり、ブロック共重合体は大きさがnmレベルなので、相転移にともなう応答の速さも好都合であった。ABAブロック共重合体を例にとると、AブロックにUCSTあるいはLCST型相挙動を示す刺激応答性高分子、Bブロックに相溶性ブロックを用いると、希薄系ではユニマーとミセルの間の転移を、濃厚系ではゾルとゲルの間の転移を温度でコントロールできることを示した(図17)44。北沢侑造博士(日本ゼオン)、小林優美博士(広栄化学)などが貢献してくれた。相分離のTcが高分子やイオン液体の構造の僅かな変化で大きく変わることに着目して、高分子中にフォトクロミック化合物を導入することを着想した。アゾベンゼン基を導入するとその光異性化状態に依存して、UCSTあるいはLCSTのTcが大きく変化することを見出した。このようなセグメントをAブロックに有するABAブロック共重合体/イオン液体系では、ある温度域で光によるユニマー・ミセル転移、ゾル・ゲル転移の誘起が可能となった。この研究にも上木博士が貢献してくれた。さらにこの現象を、光治癒材料に展開することを思いついた45。傷のある部分に紫外光をあてて、その部分のゲルを一部ゾル化して傷を埋め、その後可視光をあててゲルをゾルに戻せば光治癒ができることを示した(図18)45。馬 暁峰博士(南京林業大講師)が力学特性や治癒特性に優れる高分子の創製を行った。この研究はさらに発展して、アゾベンゼン基を高分子でなくイオン液体中に導入した系でも光による分子集合状態の可逆的変化が実現できることを、王 彩虹博士(四川大准教授)が見出してくれた46。最近のイオン液体を用いた刺激応答材料の研究に関しては、玉手亮多氏(NIMS)、橋本 慧氏(東京大学特任助教)が博士研究員として活躍してくれた。
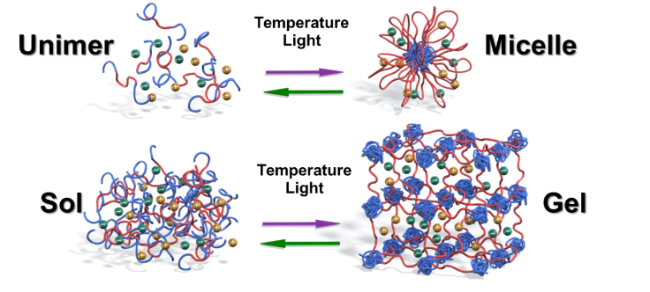
図17 イオン液体とブロック共重合体からなる自己組織体の温度や光による集合状態の可逆的制御. 44
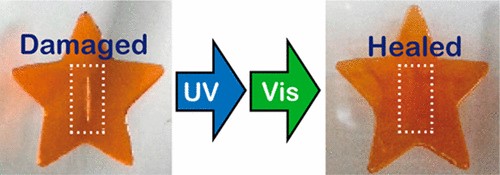
図18 アゾベンゼン基を有するブロック共重合体の光誘起ゾルゲル変化を利用した光治癒材料の提案.45
川の流れのように自分の研究領域を自分で切り拓いて来たという自負はある。また「自分の発表論文が世界の研究を動かしている」と感じられるような研究者としての醍醐味も味わうこともできた。一方で、これまで述べて来たような研究を行えたのも、一緒に研究を進めてくれた大学院生、博士研究員や共同研究者が優秀で、その力を借りて来ただけかなと思うこともある。本文中で、学生に関しては博士号を取得した方を中心に記したが、名前は記せなかったが修士までで立派な研究をしてくれた多くの学生がいる。ここに謝意を表したい。また、特にイオン液体を用いた研究に関しては、2005~2010年に実施された科研費の特定領域研究「イオン液体の科学」やさらにその後発足した本会「イオン液体研究会」の先生方と、多くの共同研究をさせて頂いた。また海外の共同研究者は、イオン液体の国際会議「Congress on Ionic Liquids(COIL)」がきっかけで知り合った方も多い。これら諸々に感謝したい。一方、大きくズームアウトして観ると、私が関係した分野では、研究を始めた頃の白川先生の研究から2019年の吉野 彰先生のノーベル化学賞の研究に繋がる潮流が、時代の要請もあり脈々と流れていた気がする。時代が研究の流れを作り、その流れに乗ってきたのかなと思う事もある。「川の流れのように」である。
References- M. Watanabe, Bull. Chem. Soc. Jpn, 2021, 94, 2739.
- M. Watanabe, Electrochemistry, 2016, 82, 642.
- H. Shirakawa, E. J. Lous, A. G. MacDiamid, C. K. Chiang, A. J. Heeger, J. Chem. Soc. Chem. Commun. 1977, 578.
- M. B. Armand, J. M. Chabagno, M. J. Duclot, In Fast Ion Transport in Solids, P. Vashishta, J. N. Mundy, G. K. Shenoy, Eds. North-Holland, New York, 1979, pp. 131-136.
- M. Watanabe, N. Ogata, Br. Polym. J. 1988, 20, 181; 渡邉正義、高分子 1993, 42, 702.
- M. Watanabe, K. Sanui, N. Ogata, T. Kobayashi, Z. Ohtaki, J. Appl. Phys. 1985, 57, 123.
- M. Watanabe, M. Itoh, K. Sanui, N. Ogata, Macromolecules 1987, 20, 569.
- R. A. Reed, L. Geng, R. W. Murray, J. Electroanal. Chem. 1986, 208, 185.
- M. Watanabe, M. L. Longmire, R. W. Murray, J. Phys. Chem. 1990, 94, 2614.
- M. Watanabe, T. T. Wooster, R. W. Murray, J. Phys. Chem. 1991, 95, 4573.
- M. J. Pinkerton, Y. Le Mest, H. Zhang, M. Watanabe, R. W. Murray, J. Am. Chem. Soc. 1990, 112, 3730.
- J. Phys. Chem. B 1999, 103, Number 20, C. Austen Angell特集号表紙.
- A. Nishimoto, K. Agehara, N. Furuya, T. Watanabe, M. Watanabe,Macromolecules 1999, 32, 1541; 渡邉正義、 高分子 1995 44, 312.
- M. Watanabe, S. Yamada, K. Sanui, and N. Ogata, J. Chem. Soc., Chem. Commun. 1993, 929.
- C. A. Angell, C. Liu, E. Sanchez, Nature 1993 , 362, 137.
- 渡邉正義、野田明宏、金子健人、川野竜司、化学と工業 2001, 54, 281.
- M. Watanabe, H. Nagasaka, N. Ogata, J. Phys. Chem. 1995, 99, 12294.
- M. Watanabe, T. Akahoshi, Y. Tabata, D. Nakayama, J. Am. Chem. Soc.1998, 120, 5577.
- S. Imabayashi, K. Ban, T. Ueki, M. Watanabe, J. Phys. Chem. B 2003, 107, 8834.
- D. Nakayama, Y. Takeoka, M. Watanabe, K. Kataoka, Angew. Chem. Int. Ed. 2003, 42, 4197.
- H. Tokuda, S. Tsuzuki, M.A.B.H. Susan, K. Hayamizu, M. Watanabe, J. Phys. Chem. B, 2006, 110, 19593.
- K. Ueno, H. Tokuda, M. Watanabe, Phys. Chem. Chem. Phys. 2010, 12, 1649.
- M.A.B.H. Susan, T. Kaneko, A. Noda, M. Watanabe, J. Am. Chem. Soc., 2005, 127, 4976.
- S. Seki, M. A. B. H. Susan, T. Kaneko, H. Tokuda, A. Noda, M. Watanabe, J. Phys. Chem. B 2005, 109, 3886.
- K. Ueno, M. Watanabe, Langmuir 2011, 27, 9105.
- M. Watanabe, M. L. Thomas, S. Zhang, K. Ueno, T. Yasuda, K. Dokko, Chem. Rev. 2017, 117, 7190.
- T. Yasuda, M. Watanabe, MRS Bull. 2013, 38, 560.
- H. Nakamoto, A. Noda, K. Hayamizu, S. Hayashi, H. Hamaguchi, M. Watanabe, J. Phys. Chem. C 2007, 111, 1541.
- S.-Y. Lee, A. Ogawa, M. Kanno, H. Nakamoto, T. Yasuda, M. Watanabe, J. Am. Chem. Soc. 2010, 132, 9764.
- S. Zhang, M. S. Miran, A. Ikoma, K. Dokko, M. Watanabe, J. Am. Chem. Soc. 2014, 136, 1690.
- R. Kawano, M. Watanabe, Chem. Commun. 2005, 2107.
- K. Takahashi, S. Sakai, H. Tezuka, Y. Hiejima, Y. Katsumura, M. Watanabe, J. Phys. Chem. B. 2007, 111, 4807.
- N. Yamanaka, R. Kawano, W. Kubo, N. Masaki, T. Kitamura, Y. Wada, M. Watanabe, S. Yanagida, J. Phys. Chem. B 2007, 111, 4763.
- R. Kawano, T. Katakabe, H. Shimosawa, M. K. Nazeeruddin, M. Grätzel, H. Matsui, T. Kitamura, N. Tanabe, M. Watanabe, Phys. Chem. Chem. Phys. 2010, 12, 1916.
- M. Watanabe, K. Dokko, K. Ueno, M. L. Thomas, Bull. Chem. Soc. Jpn. 2018, 91, 1660.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko, M. Watanabe, J. Am. Chem. Soc. 2011, 133, 13121.
- C. A. Angell, Y. Ansari, Z. Zhao, Faraday Discuss. 2012, 154, 9.
- T. Ueki, M. Watanabe, Bull. Chem. Soc. Jpn., 2012, 85, 33.
- K. Asaka, K. Oguro, Y. Nishimura, M. Mizuhata, H. Takenaka, Polym. J. 1995, 27, 436.
- S. Imaizumi, H. Kokubo, M. Watanabe, Macromolecules 2012, 45, 401.
- T. Ueki, M. Watanabe, Macromolecules 2008, 41, 3739.
- M. Matsugami, K. Fujii, T. Ueki, Y. Kitazawa, Y. Umebayashi, M. Watanabe, M. Shibayama, Anal. Sci. 2013, 29, 311.
- T. Ueki, A. Ayusawa Arai, K. Kodama, S. Kaino, N. Takata, T. Morita, K. Nishikawa, M. Watanabe, Pure & Appl. Chem. 2009, 81, 1829.
- R. Tamate, K. Hashimoto, T. Ueki, M. Watanabe, Phys. Chem. Chem. Phys. 2018, 20, 25123.
- T. Ueki, R. Usui, Y. Kitazawa, T. P. Lodge, and M. Watanabe, Macromolecules 2015, 48, 5928.
- C. Wang, K. Hashimoto, R. Tamate, H. Kokubo, M. Watanabe, Angew. Chem. Int. Ed. 2018, 57, 227.